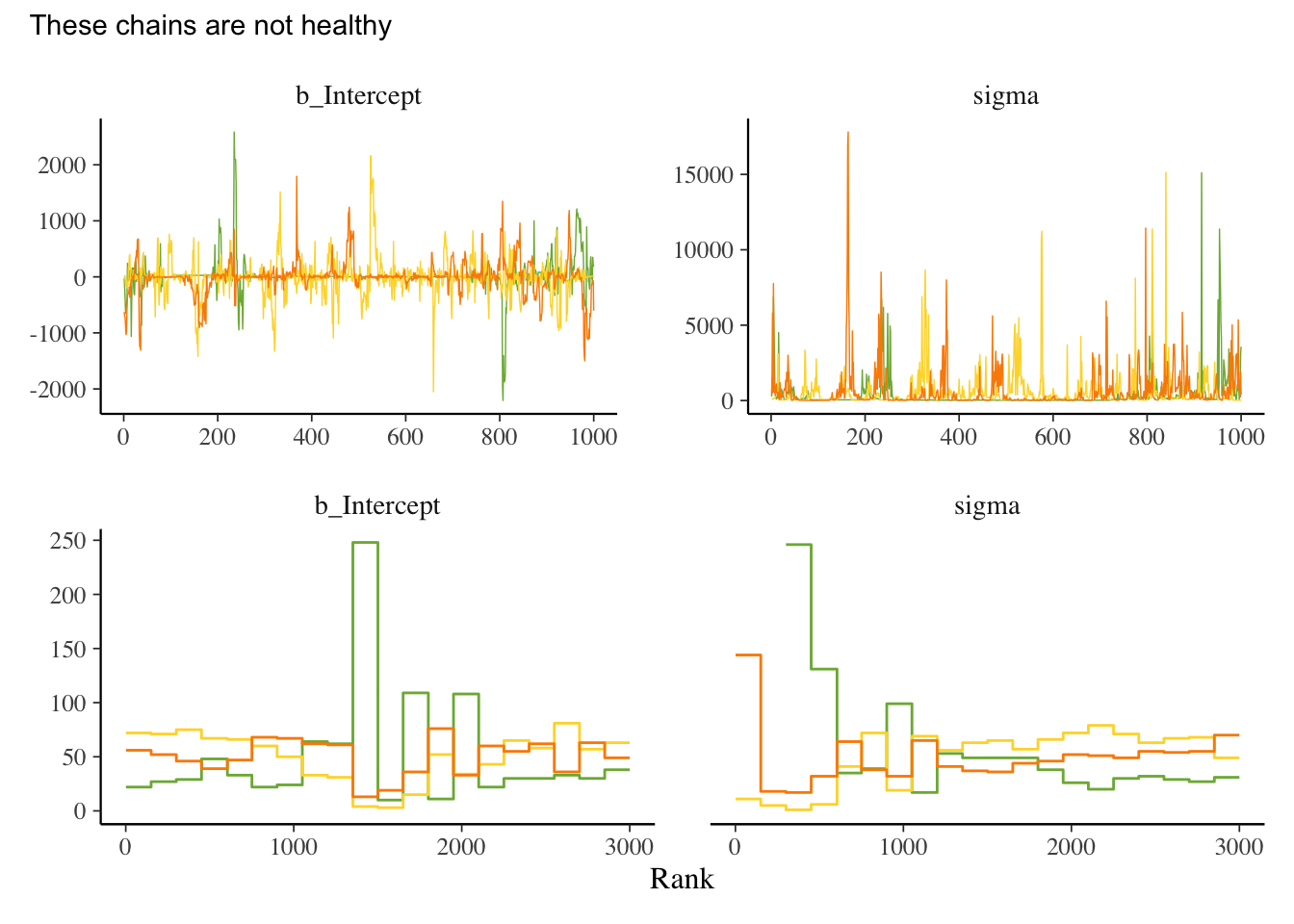
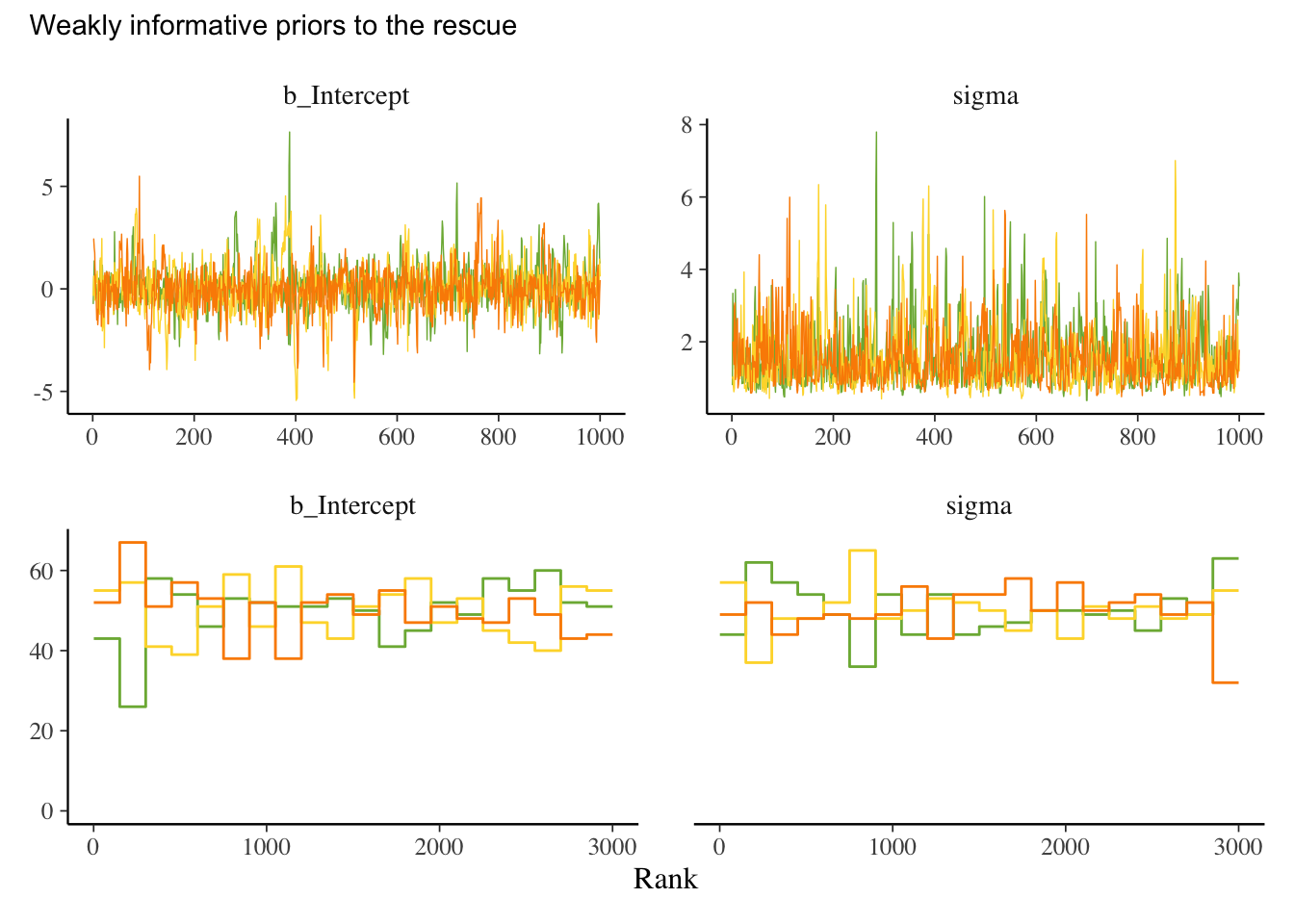
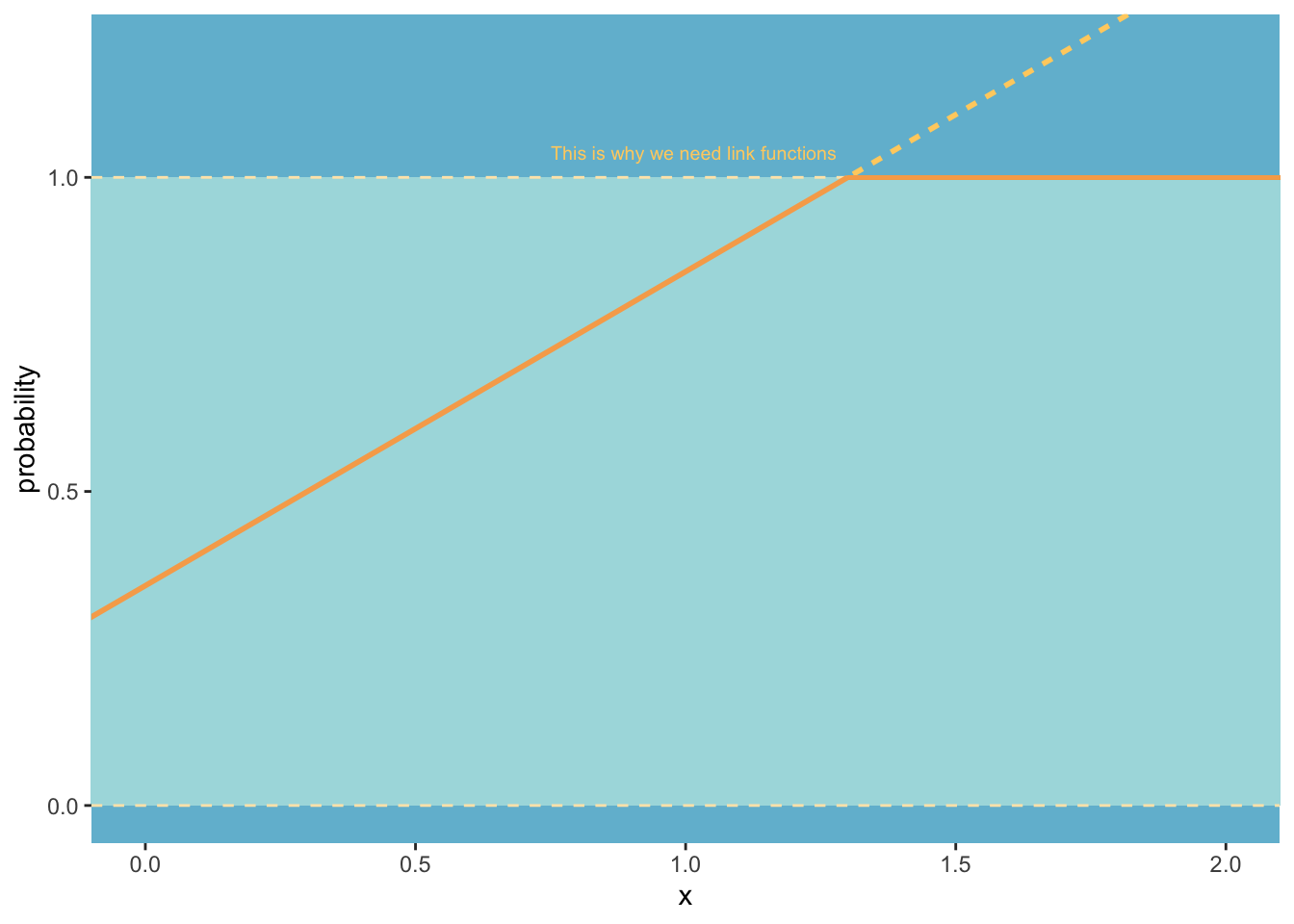
library(tidyverse) # for tidy codinglibrary(brms) # for fitting stan modelslibrary(patchwork) # for combining plotslibrary(ggdag) # drawing dagslibrary(tidybayes) # for bayesian data visualisationlibrary(bayesplot) # more bayes data vislibrary(MetBrewer) # colours library(ggrepel) # for nice text on ggplotslibrary(loo) # for information criteria
\(~\)
Lecture 1: The Golem of Prague
\(~\)
Causal inference: the attempt to understand the scientific causal model using data.
It is prediction - Knowing a cause means being able to predict the consequences of an intervention
It is a kind of imputation - Knowing a cause means being able to construct unobserved counterfactual outcomes
What if I do this types of questions.
Causal inference is intimately related to the description of populations and the design of research questions. This is because all three depend upon causal models/knowledge.
Describing populations depends upon causal inference because nealry always description depends upon the sample of the population wich you are using to predict population charactetiscs.
\(~\)
DAGs (Directed acyclic graphs)
\(~\)
Heuristic causal models that clarify scientific thinking.
We can use these to produce appropriate statistical models.
These help us think about the science before we think about the data.
Helps with questions like “what variables should we include in an analysis?”
An integral part of the course that will come up over and over again.
An example:
Lets make a function to speed up the DAG making process. We’ll use this a lot
Code
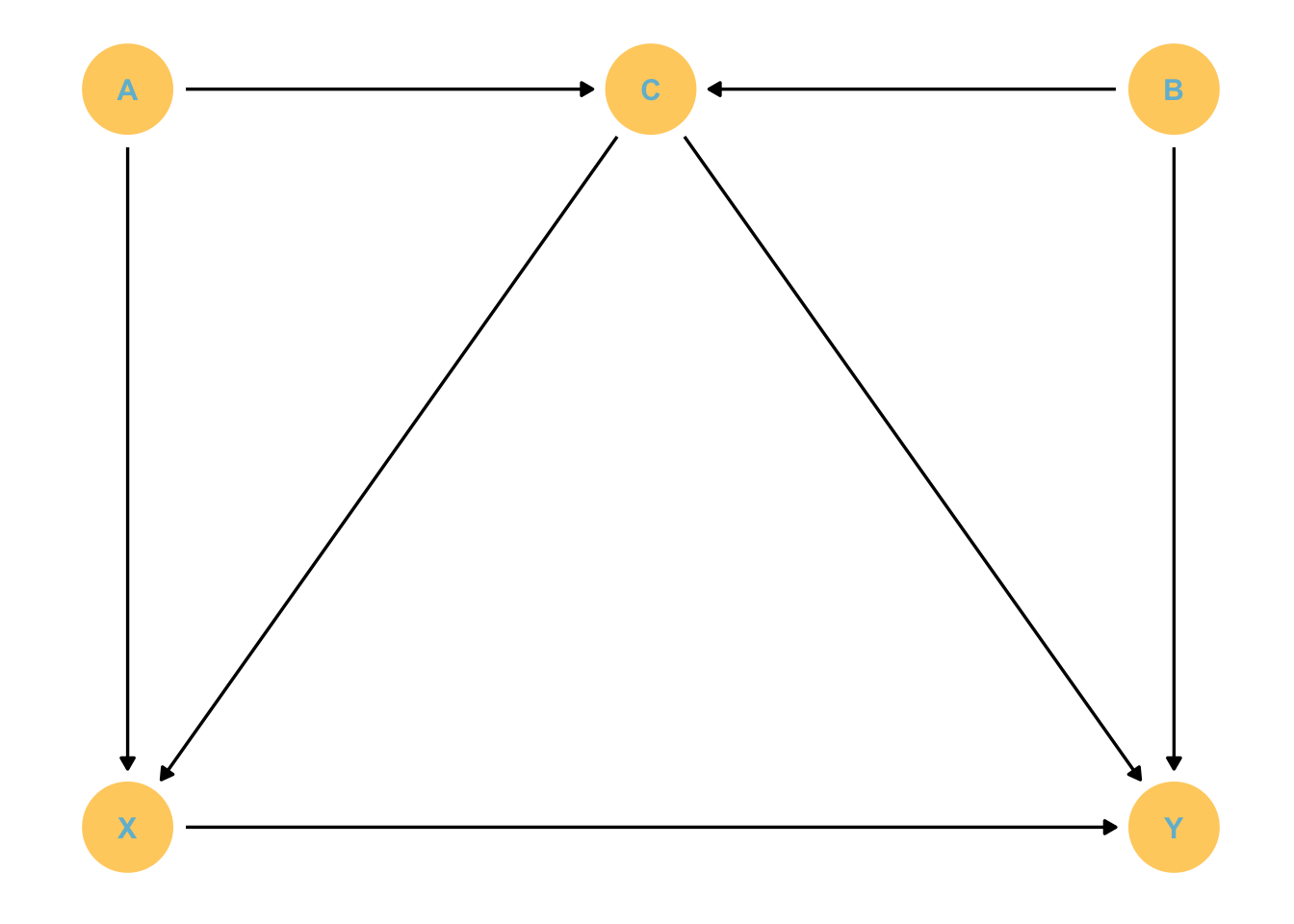
# Lets make a function to speed up the DAG making process. We'll use this a lotgg_simple_dag <-function(d) { d %>%ggplot(aes(x = x, y = y, xend = xend, yend = yend)) +geom_dag_point(color =met.brewer("Hiroshige")[4]) +geom_dag_text(color =met.brewer("Hiroshige")[7]) +geom_dag_edges() +theme_dag()}dagify( X ~ A + C, C ~ A + B, Y ~ X + C + B, coords =tibble(name =c("A", "C", "B", "X", "Y"),x =c(1, 2, 3, 1, 3),y =c(2, 2, 2, 1, 1))) %>%gg_simple_dag()
\(~\)
Golems
\(~\)
Statistical models are akin to Golems - clay robots brought to life by magic to follow specific tasks. The trouble is, they follow tasks extremely literally and are blind to their creators intent, so even those born out of the purest of intentions can cause great harm. Models are another form of robot; if we apply models within the wrong context they will not help us at all.
Ecological models are rarely designed to falsify null-hypotheses. This is because there are many possible non-null models or put another way there are no true null models in many systems. This is problematic because null models underlie how the majority of biologists do statistics!
A more appropriate question is to ask how multiple process models that we can identify are different.
Should falsify the explanatory model, not the null model. Make predictions and try and falify those. Karl Popper
\(~\)
Owls
\(~\)
Satistical modelling explanations often provide a brief introduction, then leave out many of the important details e.g. step 1: draw two circles, step 2: draw the rest of the fking owl.
We shall have a specific workflow for fitting models that we will document
Drawing the owl, or the scientific workflow can be broken down into 5 steps:
Theoretical estimand: what are you trying to do in your study in the first place?
Some scientific causal model should be identified: step 1 will be precisely defined in the context of a causal model.
Use 1 and 2 to build a statistical model.
Simulate the scientific casual model to validate that the statistical model from step 3 yields the theoretical estimand i.e. check that our stats model works.
Analyse the real data. Note that the real data are only introduced now.
\(~\)
Lecture 2: The garden of forking data
\(~\)
Globe tossing
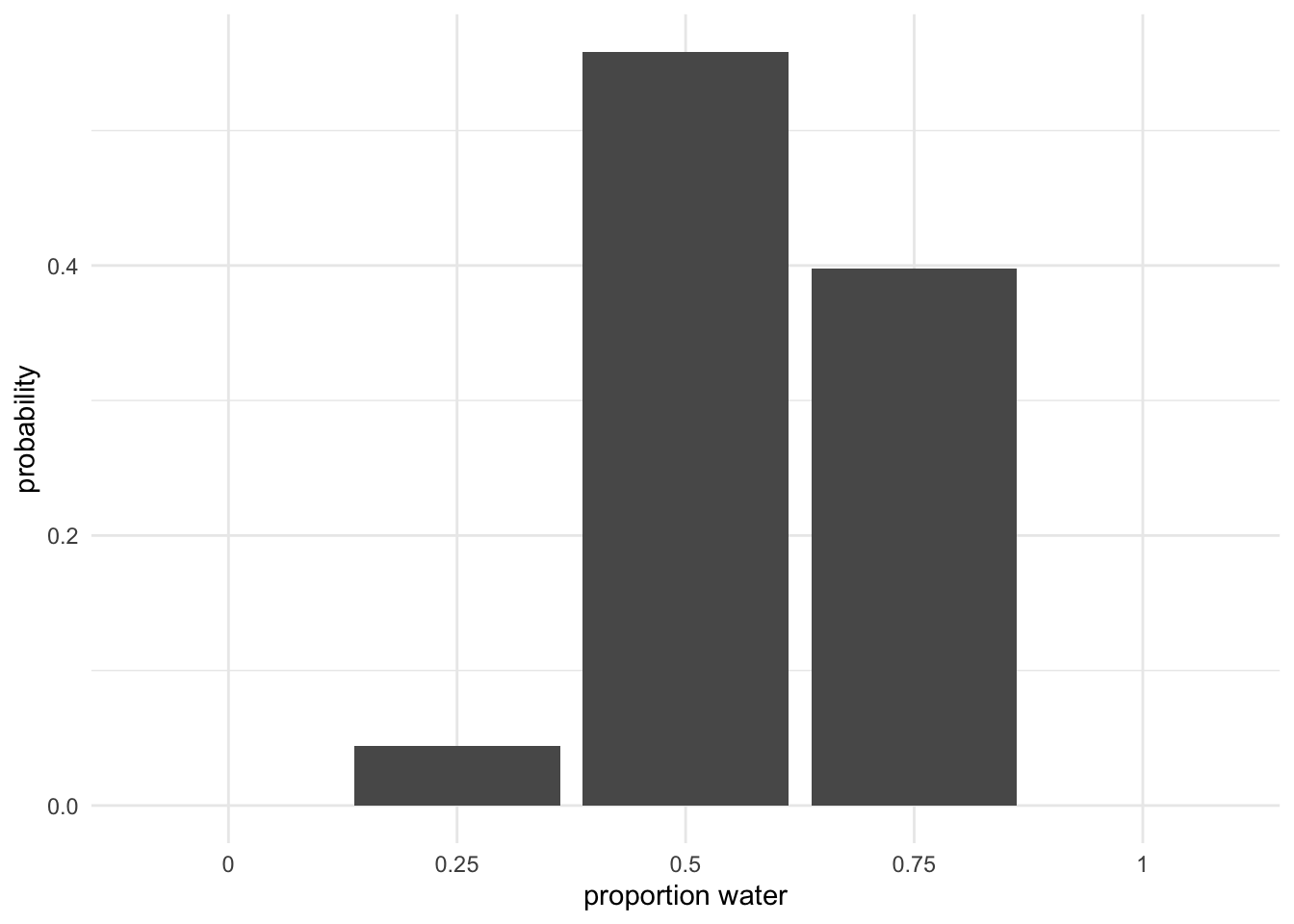
To estimate the proportion of water on planet earth, we collect data by tossing a globe 10 times and record whether our left index finger lands on land or water.
# A tibble: 10 × 1
toss
<chr>
1 l
2 w
3 l
4 l
5 w
6 w
7 w
8 l
9 w
10 w
Remember the owl workflow:
Define generative model of sample.
Design estimand.
Use 1 and 2 to build a statistical model.
Test (3) using (1).
Analyse sample and summarise.
\(~\)
Step 1
Some true proportion of water, \(p\).
We can measure this indirectly using:
\(N\): the number of globe tosses
\(W\): the number of water observations
\(L\): the number of land observations
Code
#Put dag here#N affects L and W but not the other way around# P also influences W and L
Bayesian data analysis
For each possible explanation of the data, count all the ways data can happen. Explanations with more ways to produce the data are more plausible (this is entropy).
Toss globe, probability \(p\) of observing \(W\), \(1 - p\) of \(L\).
Each toss is random because it is chaotic (there are forces that could be theoretically measured i.e. velocity, exact starting orientation, but we are not equipped to measure these in real time, so the process appears random).
Each toss is independent of one another.
What are the relative number of ways we could get the data we actually got, given the process that generates all the possible datasets that could’ve been born from the process?
\(~\)
A 4 sided globe (dice)
In Bayesian analysis: enumerate all possible outcomes.
Code
# code land as 0 and water as 1 and create possibility data# create the dataframe (tibble)d <-tibble(p_1 =0,p_2 =rep(1:0, times =c(1, 3)),p_3 =rep(1:0, times =c(2, 2)),p_4 =rep(1:0, times =c(3, 1)),p_5 =1)d %>%gather() %>%mutate(x =rep(1:4, times =5),possibility =rep(1:5, each =4)) %>%ggplot(aes(x = x, y = possibility, fill = value %>%as.character())) +geom_point(shape =21, size =9) +scale_fill_manual(values =c("white", "navy")) +scale_x_continuous(NULL, breaks =NULL) +coord_cartesian(xlim =c(.75, 4.25),ylim =c(.75, 5.25)) +theme_minimal() +theme(legend.position ="none",text =element_text(size =18))
The data: the dice is rolled three times: water, land, water.
Consider all possible answers and how they would occur.
e.g. if we hypothesise that 25% of the earth is covered by water, what are all the possible ways to produce our sample?
Code
# create a tibble of all the possibilities per marble draw, with columns position (where to appear on later figures x axis), draw (what number draw is it? and how many for each number? where to appear on figures y axis) and fill (colour of ball for figure)d <-tibble(position =c((1:4^1) /4^0, (1:4^2) /4^1, (1:4^3) /4^2),draw =rep(1:3, times =c(4^1, 4^2, 4^3)),fill =rep(c("b", "w"), times =c(1, 3)) %>%rep(., times =c(4^0+4^1+4^2)))# we wish to make a path diagram, for which we will need connecting lines, create two more tibbles for theselines_1 <-tibble(x =rep((1:4), each =4),xend = ((1:4^2) /4),y =1,yend =2)lines_2 <-tibble(x =rep(((1:4^2) /4), each =4),xend = (1:4^3) / (4^2),y =2,yend =3)# We’ve generated the values for position (i.e., the x-axis), in such a way that they’re all justified to the right, so to speak. But we’d like to center them. For draw == 1, we’ll need to subtract 0.5 from each. For draw == 2, we need to reduce the scale by a factor of 4 and we’ll then need to reduce the scale by another factor of 4 for draw == 3. The ifelse() function will be of use for that.d <- d %>%mutate(denominator =ifelse(draw ==1, .5,ifelse(draw ==2, .5/4, .5/4^2))) %>%mutate(position = position - denominator)lines_1 <- lines_1 %>%mutate(x = x - .5,xend = xend - .5/4^1)lines_2 <- lines_2 %>%mutate(x = x - .5/4^1,xend = xend - .5/4^2)# create the plot, using geom_segment to add the lines - note coord_polar() which gives th eplot a globe-like effect. scale_x_continuous and the y equivalent have been used to remove axis lables and titlesd %>%ggplot(aes(x = position, y = draw)) +geom_segment(data = lines_1,aes(x = x, xend = xend,y = y, yend = yend),size =1/3) +geom_segment(data = lines_2,aes(x = x, xend = xend,y = y, yend = yend),size =1/3) +geom_point(aes(fill = fill),shape =21, size =4) +scale_fill_manual(values =c("navy", "white")) +scale_x_continuous(NULL, limits =c(0, 4), breaks =NULL) +scale_y_continuous(NULL, limits =c(0.75, 3), breaks =NULL) +theme_minimal() +theme(panel.grid =element_blank(),legend.position ="none") +coord_polar()
Prune the number of possible outcomes down to those that are consistent with the data.
e.g. there are 3 paths consistent with our dice rolling experiment for the given data and the given hypothesis.
Code
lines_1 <- lines_1 %>%mutate(remain =c(rep(0:1, times =c(1, 3)),rep(0, times =4*3)))lines_2 <- lines_2 %>%mutate(remain =c(rep(0, times =4),rep(1:0, times =c(1, 3)) %>%rep(., times =3),rep(0, times =12*4)))d <- d %>%mutate(remain =c(rep(1:0, times =c(1, 3)),rep(0:1, times =c(1, 3)),rep(0, times =4*4),rep(1:0, times =c(1, 3)) %>%rep(., times =3),rep(0, times =12*4))) # finally, the plot:d %>%ggplot(aes(x = position, y = draw)) +geom_segment(data = lines_1,aes(x = x, xend = xend,y = y, yend = yend,alpha = remain %>%as.character()),size =1/3) +geom_segment(data = lines_2,aes(x = x, xend = xend,y = y, yend = yend,alpha = remain %>%as.character()),size =1/3) +geom_point(aes(fill = fill, alpha = remain %>%as.character()),shape =21, size =4) +# it's the alpha parameter that makes elements semitransparentscale_alpha_manual(values =c(1/10, 1)) +scale_fill_manual(values =c("navy", "white")) +scale_x_continuous(NULL, limits =c(0, 4), breaks =NULL) +scale_y_continuous(NULL, limits =c(0.75, 3), breaks =NULL) +theme_minimal() +theme(panel.grid =element_blank(),legend.position ="none") +coord_polar()
Is 3 ways to produce the sample big or small, to find out, compare with other possibilities.
e.g. lets make another conjecture - all land or all water - we have a land and water observation so there are zero ways that the all land/water hypotheses are consistent with the data.
Code
# if we make two custom functions, here, it will simplify the code within `mutate()`, belown_water <-function(x){rowSums(x =="W")}n_land <-function(x){rowSums(x =="L")}t <-# for the first four columns, `p_` indexes positiontibble(p_1 =rep(c("L", "W"), times =c(1, 4)),p_2 =rep(c("L", "W"), times =c(2, 3)),p_3 =rep(c("L", "W"), times =c(3, 2)),p_4 =rep(c("L", "W"), times =c(4, 1))) %>%mutate(`roll 1: water`=n_water(.),`roll 2: land`=n_land(.),`roll 3: water`=n_water(.)) %>%mutate(`ways to produce`=`roll 1: water`*`roll 2: land`*`roll 3: water`)t %>% knitr::kable()
p_1
p_2
p_3
p_4
roll 1: water
roll 2: land
roll 3: water
ways to produce
L
L
L
L
0
4
0
0
W
L
L
L
1
3
1
3
W
W
L
L
2
2
2
8
W
W
W
L
3
1
3
9
W
W
W
W
4
0
4
0
Unglamorous basis of applied probability: Things that can happen more ways are more plausible.
This is Bayesian inference. Given a set of assumptions (hypotheses) the number of ways the numbers could have occurred accoridng to those assumptions (hypotheses) is the posterior probability distribution.
But we don’t really have enough evidence to be confident in a prediction. So lets roll the dice again. In bayes world a process called Bayesian updating exists, that saves you from running the draws again, in favour of just updating previous counts (or data). Bayesian updating is simple multiplication e.g. we roll another water, for the 3 water hypothesis there are 3 paths for this to occur so we multiply 9 x 3, resulting in 27 possible paths consistent with the new dataset.
Eventually though, the garden gets really big. This is where your computer comes in and it starts to make more sense to work with probabilities rather than counts.
sim_globe <-function(p =0.7, N =9) {sample(c("W", "L"), size = N, prob =c(p, 1-p), replace =TRUE)}# W and L are the possible observations# N is number of tosses# prob is the probabilities of water and land occurring
The simulation does this:
Code
sim_globe()
[1] "W" "W" "W" "W" "L" "W" "W" "W" "W"
Now lets test it on extreme settings
e.g. all water
Code
sim_globe(p=1, N =11)
[1] "W" "W" "W" "W" "W" "W" "W" "W" "W" "W" "W"
Looks good
We can also test how close the proportion of water produced in the simulation is to the specified p
Code
sum(sim_globe(p=0.5, N =1e4) =="W") /1e4
[1] 0.4986
Also looks good.
So based upon our generative model:
Ways for \(p\) to produce $W, L = (4p)^W * (4 - 4p)^L $
Code
# function to compute posterior distirbutioncompute_posterior <-function(the_sample, poss =c(0, 0.25, 0.5, 0.75, 1)) { W <-sum(the_sample =="W") # the number of W observed L <-sum(the_sample =="L") # the number of L observed ways <-sapply(poss, function(q) (q*4)^W * ((1-q)*4)^L) post <- ways/sum(ways)#bars <- sapply(post, function(q) make_bar(q))tibble(poss, ways, post =round(post, 3))#, bars)}
We can then simulate the experiment many times with sim_globe
This allows us to check that our model is doing what we think it is.
\(~\)
An infinite number of possibilties
Globes aren’t dice. There are an infinite number of possible proportions of the earth covered in water.
To get actual infinity we can turn to math.
The relative number of ways that nay value of \(p\) could produce any sample of W and L observations. This is a well know distribution called the binomial distribution
\[p^W(1-p)^L \]
The only trick is normalising to make it a probability. A little calculus is needed (this produces the beta distirbution:
Note the normalising constant \(\frac{(W + L)!}{W!L!}\)
We can use the binomial sampling formula to give us the number of paths through the garden of forking data for this particular problem. That is given some value of P (akin to some number of blue marbles in the bag), the number of ways to see W and L can be worked out using the expression above.
We can use R to calculate this, assuming 6 globe tosses that landed on water out of 10 possible tosses and that P = 0.7:
Code
dbinom(6, 10, 0.7)
[1] 0.2001209
This is the relative number of ways that 6 out of 10 can happen given a value of 0.7 for p. For this to be meaningful, we need to work out a probability value for many other values of P. This gives our probability distribution.
Lets plot how bayesian updating works, as we add observations
Code
# add the cumulative number of trials and successes for water to the dataframe.globe_toss_data <- globe_toss_data %>%mutate(n_trials =1:10, n_success =cumsum(toss =="w"))# Struggling to follow this code, awesome figure produced thoughsequence_length <-50globe_toss_data %>%expand(nesting(n_trials, toss, n_success),p_water =seq(from =0, to =1, length.out = sequence_length)) %>%group_by(p_water) %>%mutate(lagged_n_trials =lag(n_trials, k =1),lagged_n_success =lag(n_success, k =1)) %>%ungroup() %>%mutate(prior =ifelse(n_trials ==1, .5,dbinom(x = lagged_n_success,size = lagged_n_trials,prob = p_water)),likelihood =dbinom(x = n_success,size = n_trials,prob = p_water),strip =str_c("n = ", n_trials) ) %>%# the next three lines allow us to normalize the prior and the likelihood, # putting them both in a probability metricgroup_by(n_trials) %>%mutate(prior = prior /sum(prior),likelihood = likelihood /sum(likelihood)) %>%# plot timeggplot(aes(x = p_water)) +geom_line(aes(y = prior), linetype =2) +geom_line(aes(y = likelihood)) +scale_x_continuous("proportion water", breaks =c(0, 0.5, 1)) +scale_y_continuous("plausibility", breaks =NULL) +theme(panel.grid =element_blank()) +facet_wrap(~n_trials, scales ="free_y")
Posterior is continually updated as data points are added.
Each posterior is simply a multiplication of a set of diagonal lines, like that shown in the first panel. Whether the slope of the line is pos or neg depends on whether the observation is land or water.
Every posterior is a prior for the next observation, but the data order doesn’t make a difference, in that the posterior will always be the same for a given dataset. BUT this assumption can be violated if each observation is not independent.
Sample size is embodied within the shape of the posterior. Already been accounted for.
Some big things to grasp about bayes
No minimum sample size, because we have something called a prior. Note that estimation sucks with small samples, but that’s good! The model doesn’t draw too much from too little.
Shape of distribution embodies sample size
There are no point estimates e.g. means or medians. Everything is a distribution. You will never need to bootstrap again.
There is no one true interval - they just communicate the shape of the posterior distribution. No magical number e.g. 95%.
\(~\)
From posterior to prediction (brms time)
Here is a nice point to introduce brms the main modelling package that I use.
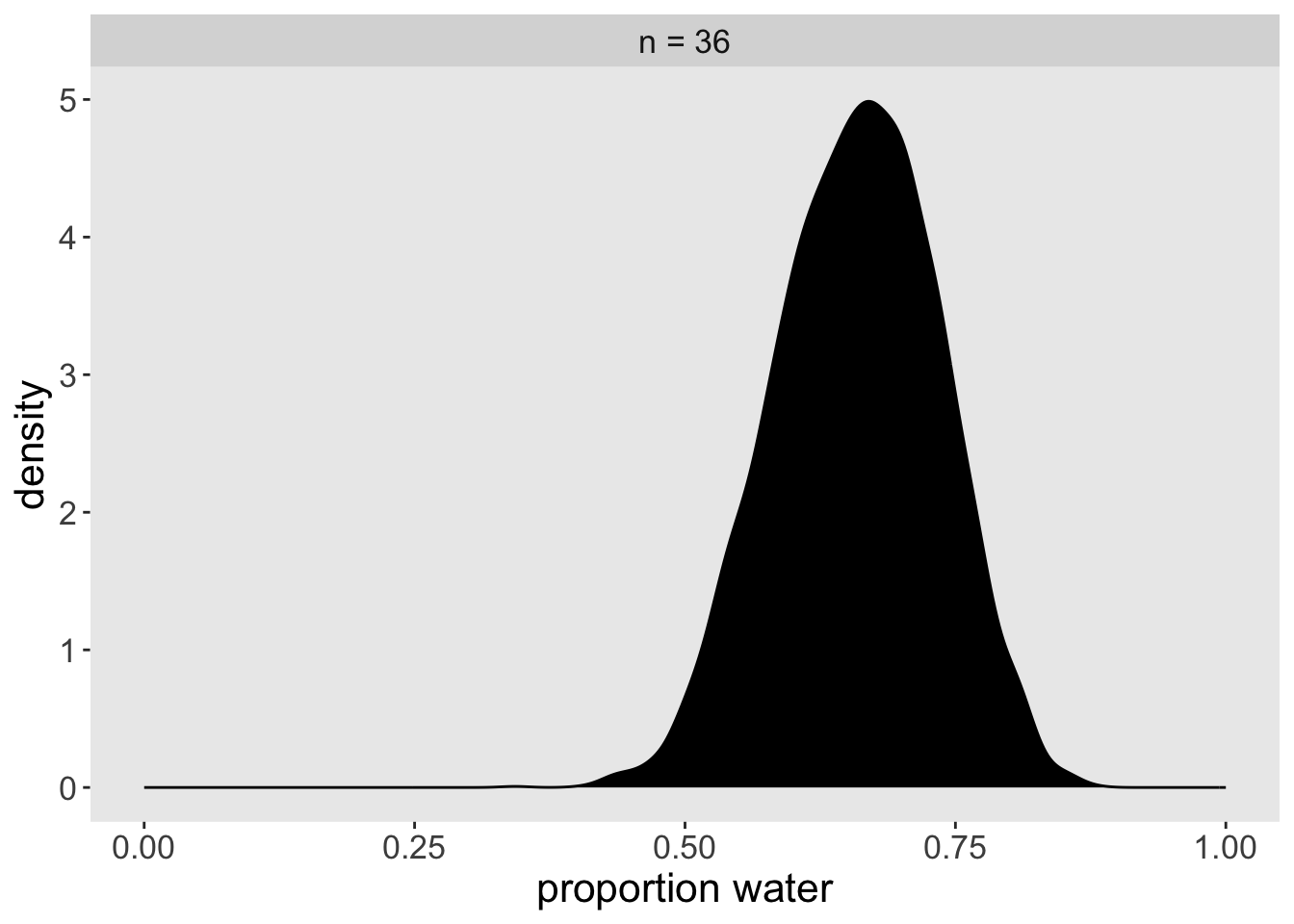
Let’s fit a globe tossing model, where we observe 24 water observations from 36 tosses.
Code
b1.1<-brm(data =list(w =24), family =binomial(link ="identity"), w |trials(36) ~0+ Intercept,#prior(beta(1, 1), class = b, lb = 0, ub = 1),seed =2,file ="fits/b02.01")b1.1
Family: binomial
Links: mu = identity
Formula: w | trials(36) ~ 0 + Intercept
Data: list(w = 24) (Number of observations: 1)
Draws: 4 chains, each with iter = 2000; warmup = 1000; thin = 1;
total post-warmup draws = 4000
Population-Level Effects:
Estimate Est.Error l-95% CI u-95% CI Rhat Bulk_ESS Tail_ESS
Intercept 0.66 0.08 0.51 0.80 1.00 1681 1947
Draws were sampled using sampling(NUTS). For each parameter, Bulk_ESS
and Tail_ESS are effective sample size measures, and Rhat is the potential
scale reduction factor on split chains (at convergence, Rhat = 1).
There’s a lot going on in that output. For now, focus on the ‘Intercept’ line. The intercept of a typical regression model with no predictors is the same as its mean. In the special case of a model using the binomial likelihood, the mean is the probability of a 1 in a given trial.
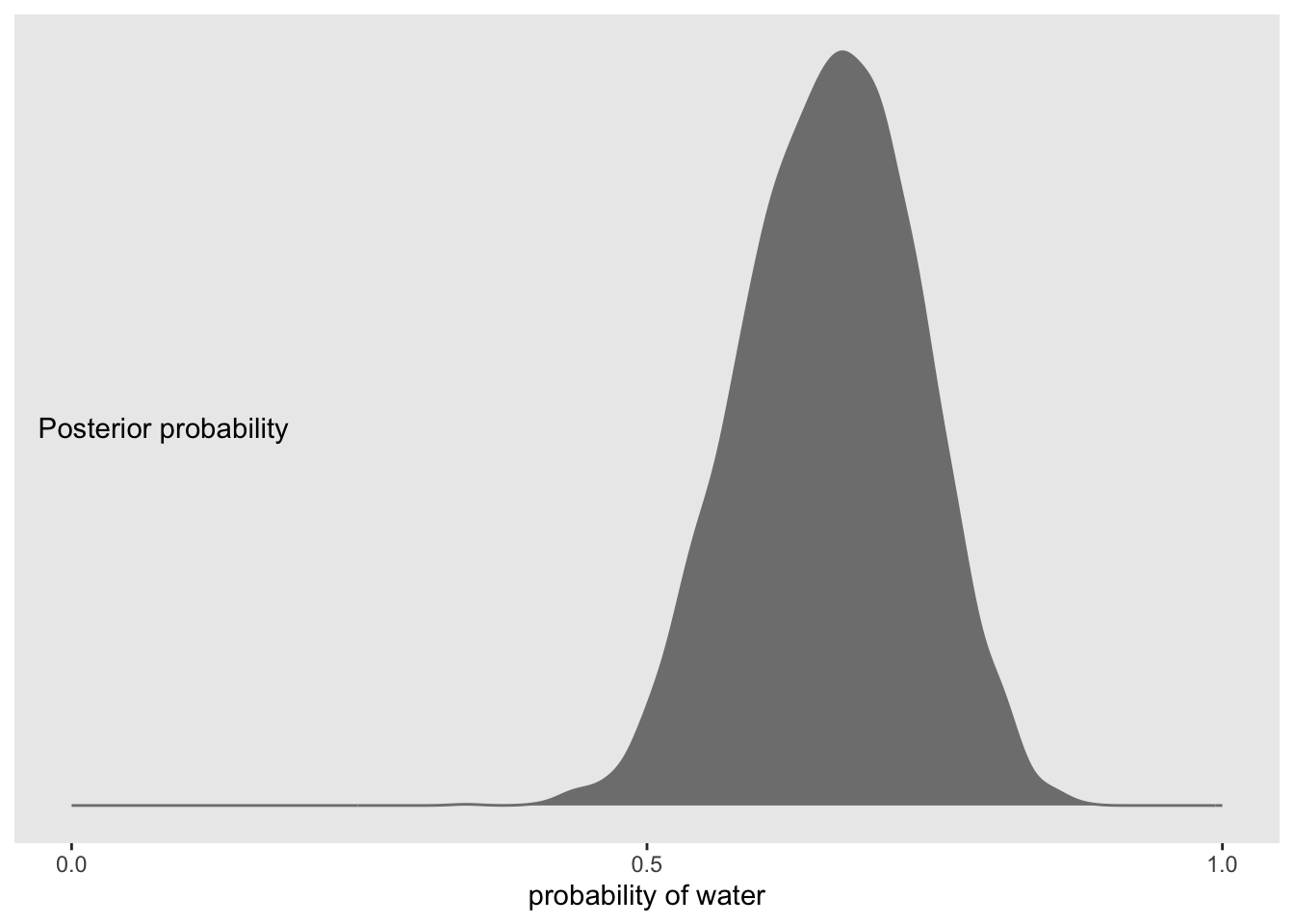
To use the posterior, we can sample from it using the as_draws_df function.
f %>%ggplot(aes(x = proportion)) +geom_density(fill ="grey50", color ="grey50") +annotate(geom ="text", x = .08, y =2.5,label ="Posterior probability") +scale_x_continuous("probability of water",breaks =c(0, .5, 1),limits =0:1) +scale_y_continuous(NULL, breaks =NULL) +theme(panel.grid =element_blank())
Strikingly similar (well, exactly the same).
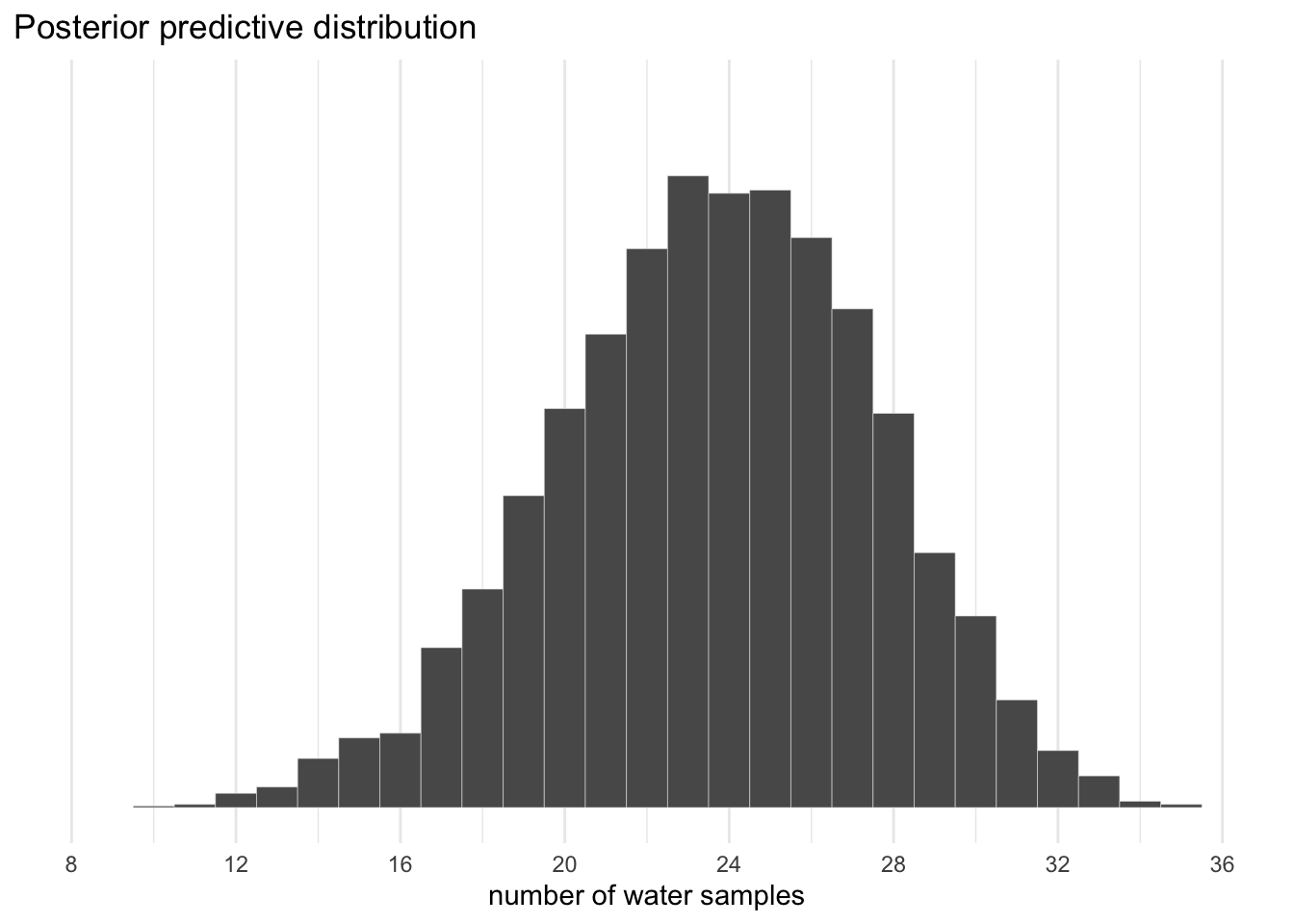
We can use this distribution of probabilities to predict histograms of water counts.
Code
# the simulationset.seed(3) # so we get the same result every timef <- f %>%mutate(w =rbinom(n(), size =36, prob = proportion))# the plotf %>%ggplot(aes(x = w)) +geom_histogram(binwidth =1, center =0,color ="grey92", size =1/10) +scale_x_continuous("number of water samples", breaks =0:40*4) +scale_y_continuous(NULL, breaks =NULL, limits =c(0, 450)) +ggtitle("Posterior predictive distribution") +coord_cartesian(xlim =c(8, 36)) +theme_minimal()
\(~\)
Lecture 3: Geocentric models
\(~\)
It is entirely possible for a completely false model to make very accurate predictions.
Geocentrism is Mcelreath’s example: the idea that the planets orbit the earth and the model behind it can very accurately predict where Mars will be in the nights sky, including when it will be in retrograde (appearing to orbit in the opposite direction).
However, just because a model allows us to make accurate predictions does not mean it is correct. We now know that the planets orbit the sun, and this more correct model can also explain mar’s apparent retrograde movement.
The relevance of this anecdote is that prediction without explanation is thwart with danger, but at the same time models like the geocentric can be very helpful.
Linear regression is similar.
They are often descriptively accurate but mechanistically wrong.
Few things in nature are perfectly linear as is assumed.
They are useful but we must remember their limits
\(~\)
What are linear models
\(~\)
Simple statistical golems that model the mean and the variance of a variable. That’s it.
The mean is sum weighted sum of other variables. As those variables change, the mean and variance changes.
Anovas, Ancovas, t-tests are all linear models.
The normal distribution
Counts up all the ways the observations can happen given a set of assumptions.
Given some mean and variance the normal distribution gives you the relative number of ways the data can appear.
The normal distribution is the norm because:
It is very common in nature.
It’s easy to calculate
Very conservative assumptions (spreads probability out more than any other distribution, reducing the risk of mistake, at the expense of accuracy)
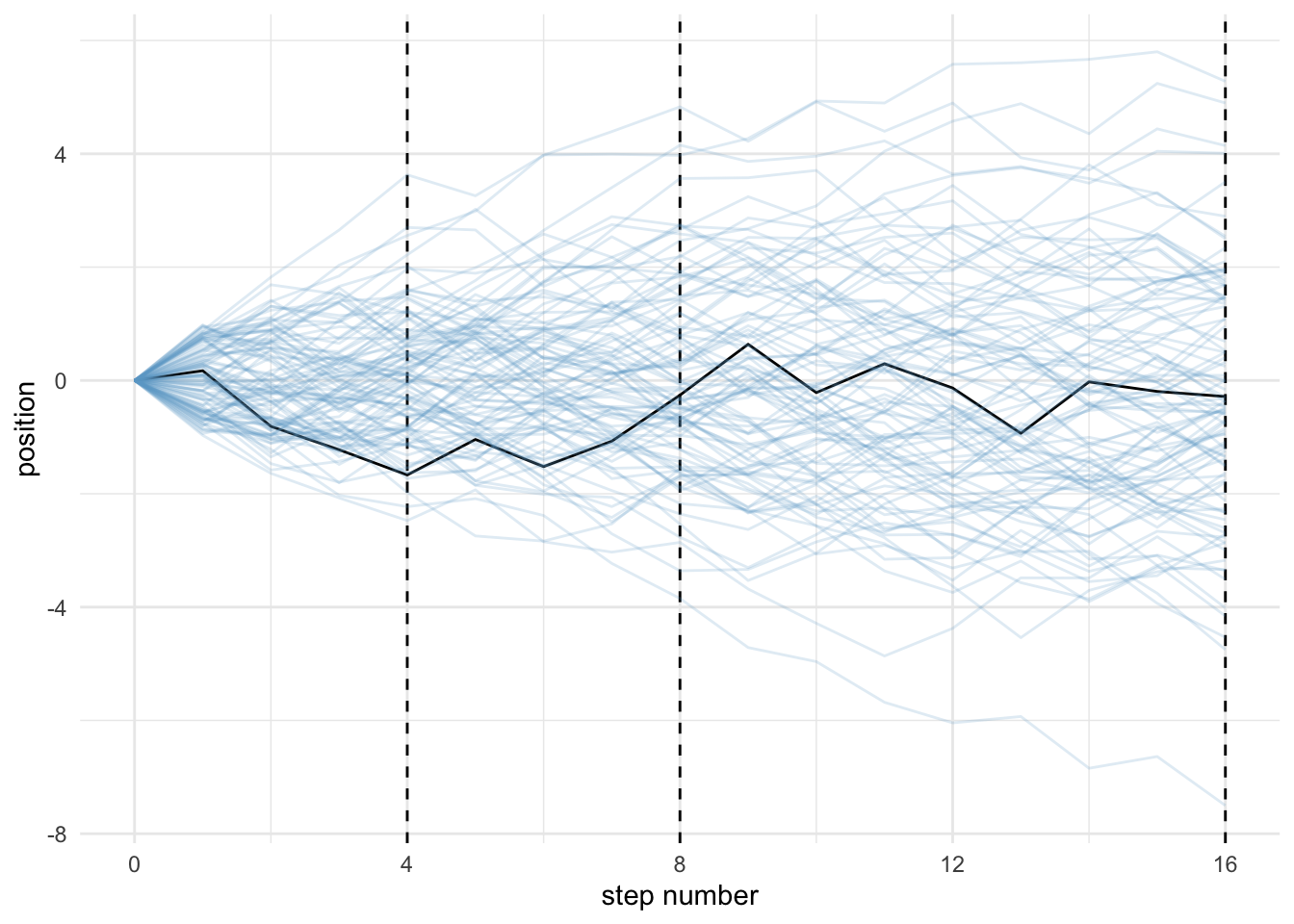
To understand why the normal distribution is so common consider a soccer pitch mid-line, and individuals flipping coins that dictate whether they should step to the left or the right.
Many individuals remain close to the line, while a few move towards the extremes. Simply, there are more ways to get a difference of 0 than there is any other result. This creates a bell curve and summarises processes to mean and variance. The path of one individual is shaded black in the figure below.
Code
# lets simulate this scenarioset.seed(4)pos <-replicate(100, runif(16, -1, 1)) %>%# this is the simas_tibble() %>%rbind(0, .) %>%# add a row of zeros above simulation resultsmutate(step =0:16) %>%# creates a step columngather(key, value, -step) %>%# convert data to long formatmutate(person =rep(1:100, each =17)) %>%# person IDs added# the next lines allow us to make cumulative sums within each persongroup_by(person) %>%mutate(position =cumsum(value)) %>%ungroup() # allows more data manipulationggplot(data = pos,aes(x = step, y = position, group = person)) +geom_vline(xintercept =c(4, 8, 16), linetype =2) +geom_line(aes(colour = person <2, alpha = person <2)) +scale_colour_manual(values =c("skyblue3", "black")) +scale_alpha_manual(values =c(1/5, 1)) +scale_x_continuous("step number", breaks =c(0, 4, 8, 12, 16)) +theme_minimal() +theme(legend.position ="none")
1. In this lecture we are going to focus on describing the association between height and weight in adults.
2. Scientific model: how does height affect weight?
Height influences weight, but not the other way around. Height is causal.
\(W = f(H)\) this means that weight is some function of height.
Code
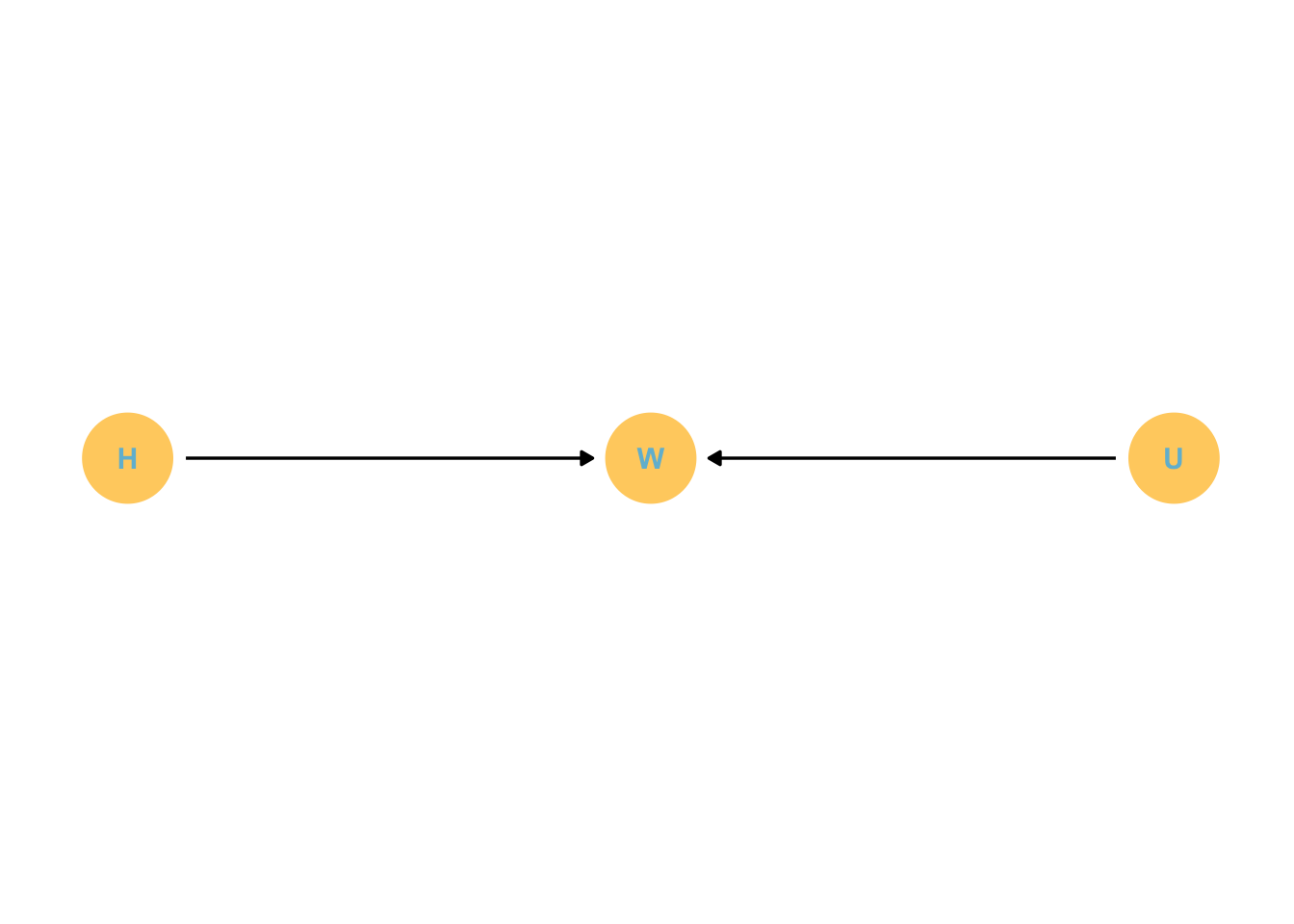
dagify( W ~ H + U, coords =tibble(name =c("H", "W", "U"),x =c(1, 2, 3),y =c(1, 1, 1))) %>%gg_simple_dag()
Note the \(U\) in the DAG - an unobserved influence on weight.
Build a generative model
\(W = \beta H + U\)
Lets simulate the data
Code
sim_weight <-function(H, b, sd){ U <-rnorm(length(H), 0, sd) W <- b*H + Ureturn(W)}
Run the simulation and plot. We’ll need values for heights, a \(\beta\) value and some value of sd for weight in kgs
Code
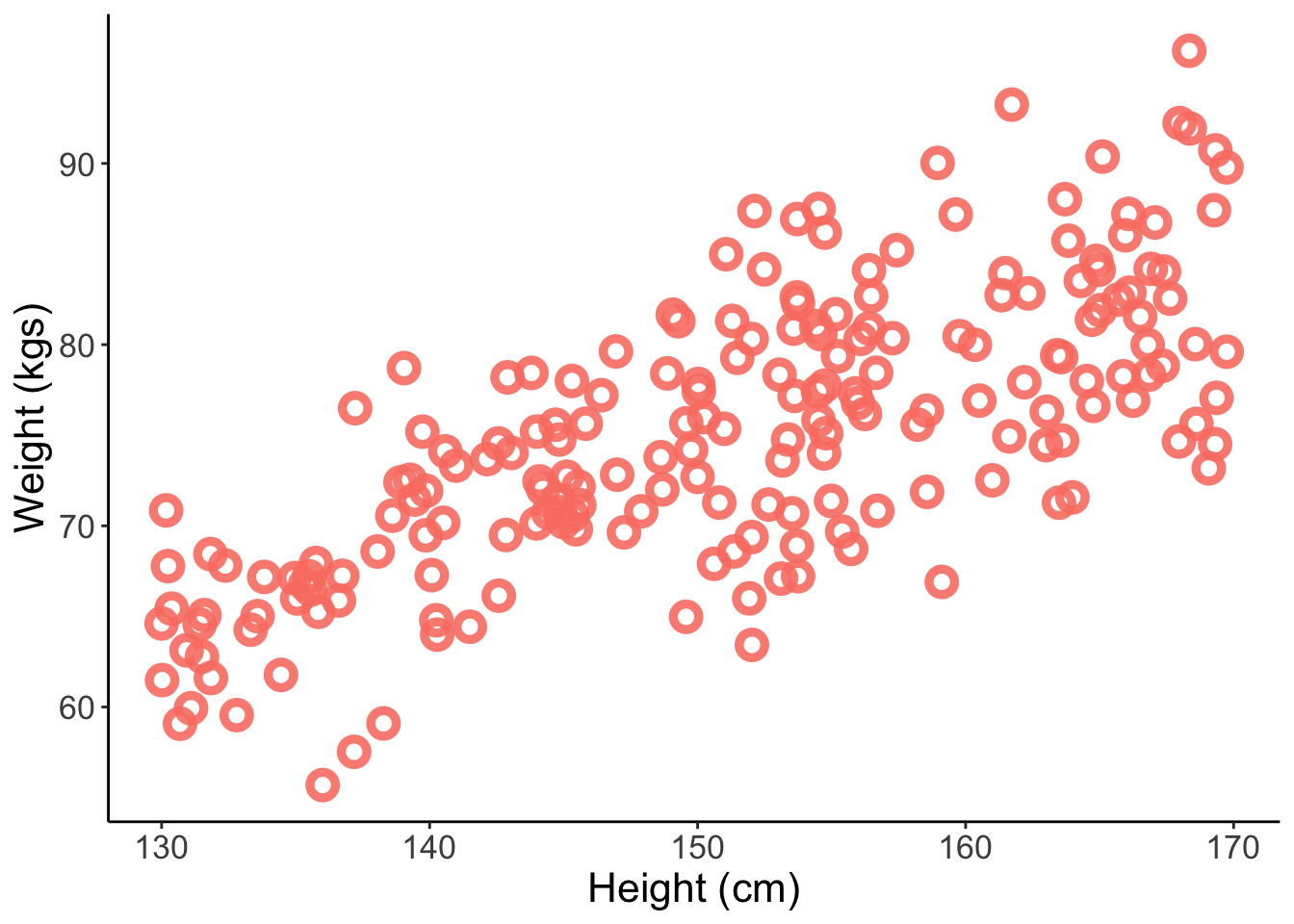
sim_data <-tibble(H =runif(200, min =130, max =170)) %>%mutate(W =sim_weight(H, b =0.5, sd =5))# plotsim_data %>%ggplot(aes(x = H, y = W)) +geom_point(color ="salmon", shape =1.5, stroke =2.5, size =3, alpha =0.9) +theme_classic() +labs(x ="Height (cm)", y ="Weight (kgs)") +theme(text =element_text(size =16))
Describing our model
\(W_{i} = \beta H_{i} + U_i\) is our equation for expected weight
\(U_{i} = Normal(0,\sigma)\) is the Gaussian error with sd \(\sigma\)
\(H_{i} =\) Uniform\((130, 170)\) means that all values are equally likely for height between 130-170
\(i\) is an index and here represents individuals in the dataset
= indicates a deterministic relationship
~ indicates that something is “distributed as”
\(~\)
Build estimator
A linear model:
\(E(W_i|H_i) = \alpha + \beta H_{i}\)
\(\alpha\) = the intercept
\(\beta\) = the slope
Our estimator:
\(W_{i}\)~\(Normal(u_{i},\sigma)\)
\(u_{i}\)~\(\alpha + \beta H_{i}\)
In words: \(W\) is distributed normally with mean that is a linear function of H
Priors
We can specify these to be very helpful. We simply want to design priors to stop the model hallucinating impossible outcomes e.g. negative weights.
Priors should express scientific knowledge, but softly. This is because the real process in nature is different to what we imagined and there needs to be room for this.
Some basic things we know about weight:
When height is zero, weight should be zero.
\(\alpha\) ~ Normal(0, 10) will achieve this
Weight increases with height in humans on average. So \(\beta\) should be positive
Weight in kgs is less than height in cms, so \(\beta\) should be less than 1
\(\beta\) ~ Uniform(0, 1) will achieve this
\(\sigma\) must be positive
\(\sigma\) ~ Uniform(0, 10)
Lets plot these priors
Code
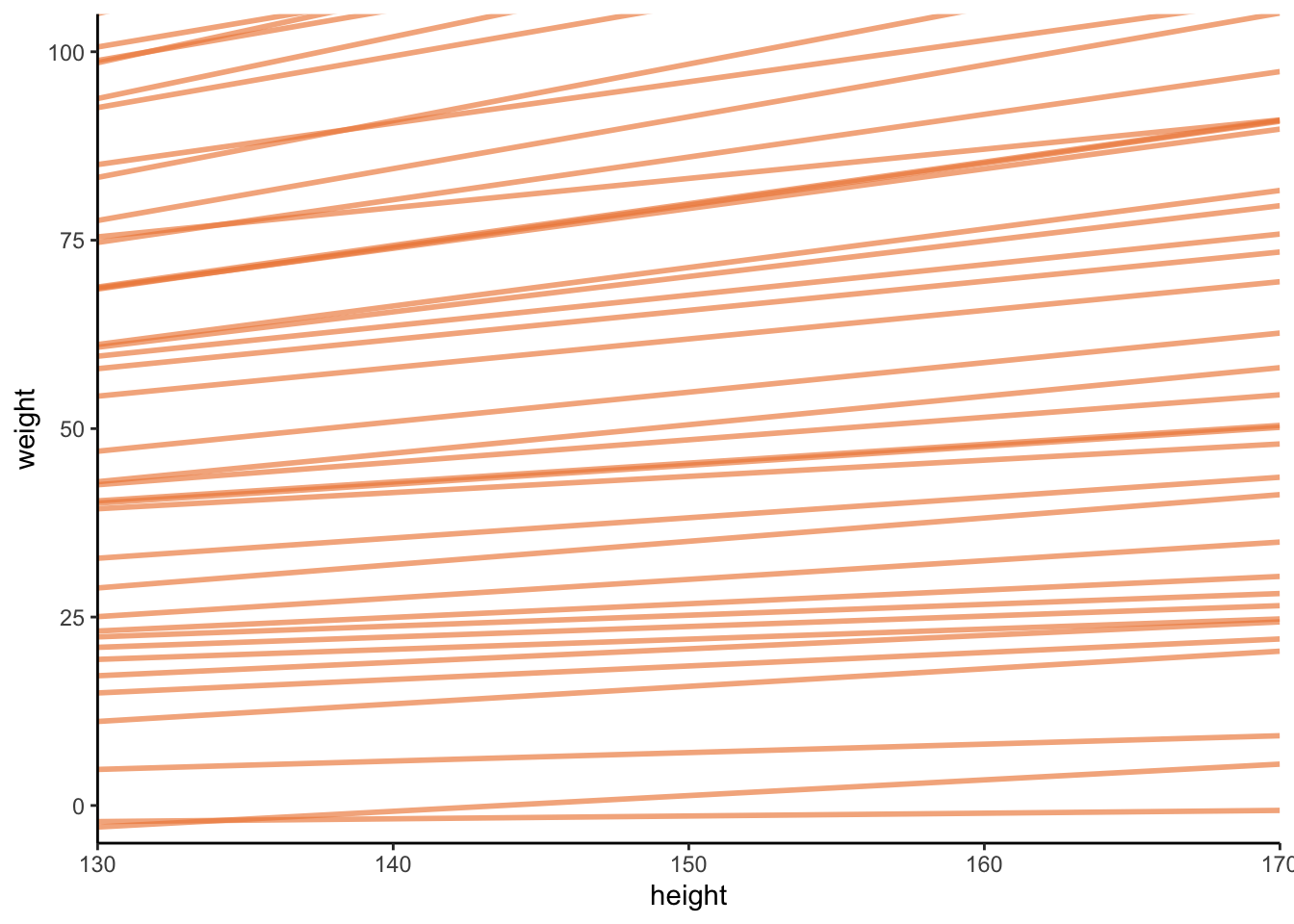
n_lines <-50tibble(n =1:n_lines,a =rnorm(n_lines, 0, 10),b =runif(n_lines, 0, 1)) %>%expand(nesting(n, a, b), height =130:170) %>%mutate(weight = a + b * (height)) %>%# plotggplot(aes(x = height, y = weight, group = n)) +geom_line(alpha =1/1.5, linewidth =1, colour =met.brewer("Hiroshige")[2]) +scale_x_continuous(expand =c(0, 0)) +coord_cartesian(ylim =c(0, 100),xlim =c(130, 170)) +theme_classic()
Woah, ok the slope looks ok, but the intercept is wild.
This is because we set a very high sd value for \(\alpha\)
We can fix this, but for a problem like this, the data will overwhelm the prior.
\(~\)
Back to the owl: brms time
Lets validate our model
Let simulate again with our sim_weight() function
Code
sim_data_100 <-tibble(H =runif(100, min =130, max =170)) %>%mutate(W =sim_weight(H, b =0.5, sd =5))
Fit the model
Code
weight_synthetic_model <-brm(W ~1+ H,family = gaussian,data = sim_data_100,prior =c(prior(normal(0, 10), class = Intercept),prior(uniform(0, 1), class = b, lb =0, ub =1),prior(uniform(0, 10), class = sigma, lb =0, ub =10)),chains =4, cores =4, iter =6000, warmup =2000, seed =1,file ="fits/weight_synthetic_model")
Get the model summary
Code
weight_synthetic_model
Family: gaussian
Links: mu = identity; sigma = identity
Formula: W ~ 1 + H
Data: sim_data_100 (Number of observations: 100)
Draws: 4 chains, each with iter = 6000; warmup = 2000; thin = 1;
total post-warmup draws = 16000
Population-Level Effects:
Estimate Est.Error l-95% CI u-95% CI Rhat Bulk_ESS Tail_ESS
Intercept 12.28 6.51 -0.70 25.06 1.00 15740 11165
H 0.42 0.04 0.34 0.51 1.00 15754 11351
Family Specific Parameters:
Estimate Est.Error l-95% CI u-95% CI Rhat Bulk_ESS Tail_ESS
sigma 4.91 0.35 4.28 5.67 1.00 14738 10608
Draws were sampled using sampling(NUTS). For each parameter, Bulk_ESS
and Tail_ESS are effective sample size measures, and Rhat is the potential
scale reduction factor on split chains (at convergence, Rhat = 1).
Right so, H is close to 0.5 and sigma is very close to 4.91. These aren’t perfect because we have a smallish sample and relatively wild priors.
Family: gaussian
Links: mu = identity; sigma = identity
Formula: weight ~ 1 + height
Data: Kalahari_adults (Number of observations: 346)
Draws: 4 chains, each with iter = 6000; warmup = 2000; thin = 1;
total post-warmup draws = 16000
Population-Level Effects:
Estimate Est.Error l-95% CI u-95% CI Rhat Bulk_ESS Tail_ESS
Intercept -51.66 4.55 -60.54 -42.77 1.00 16472 11740
height 0.63 0.03 0.57 0.68 1.00 16478 11912
Family Specific Parameters:
Estimate Est.Error l-95% CI u-95% CI Rhat Bulk_ESS Tail_ESS
sigma 4.27 0.16 3.97 4.60 1.00 15291 11571
Draws were sampled using sampling(NUTS). For each parameter, Bulk_ESS
and Tail_ESS are effective sample size measures, and Rhat is the potential
scale reduction factor on split chains (at convergence, Rhat = 1).
\(~\)
OBEY THE LAW
\(~\)
Law 1. Parameters are not independent of one another and cannot always be interpreted independently. They all act simultaneously on predictors (think about this from the context of the fitted() function).
Instead we can push out posterior predictions from the model and describe/interpret those.
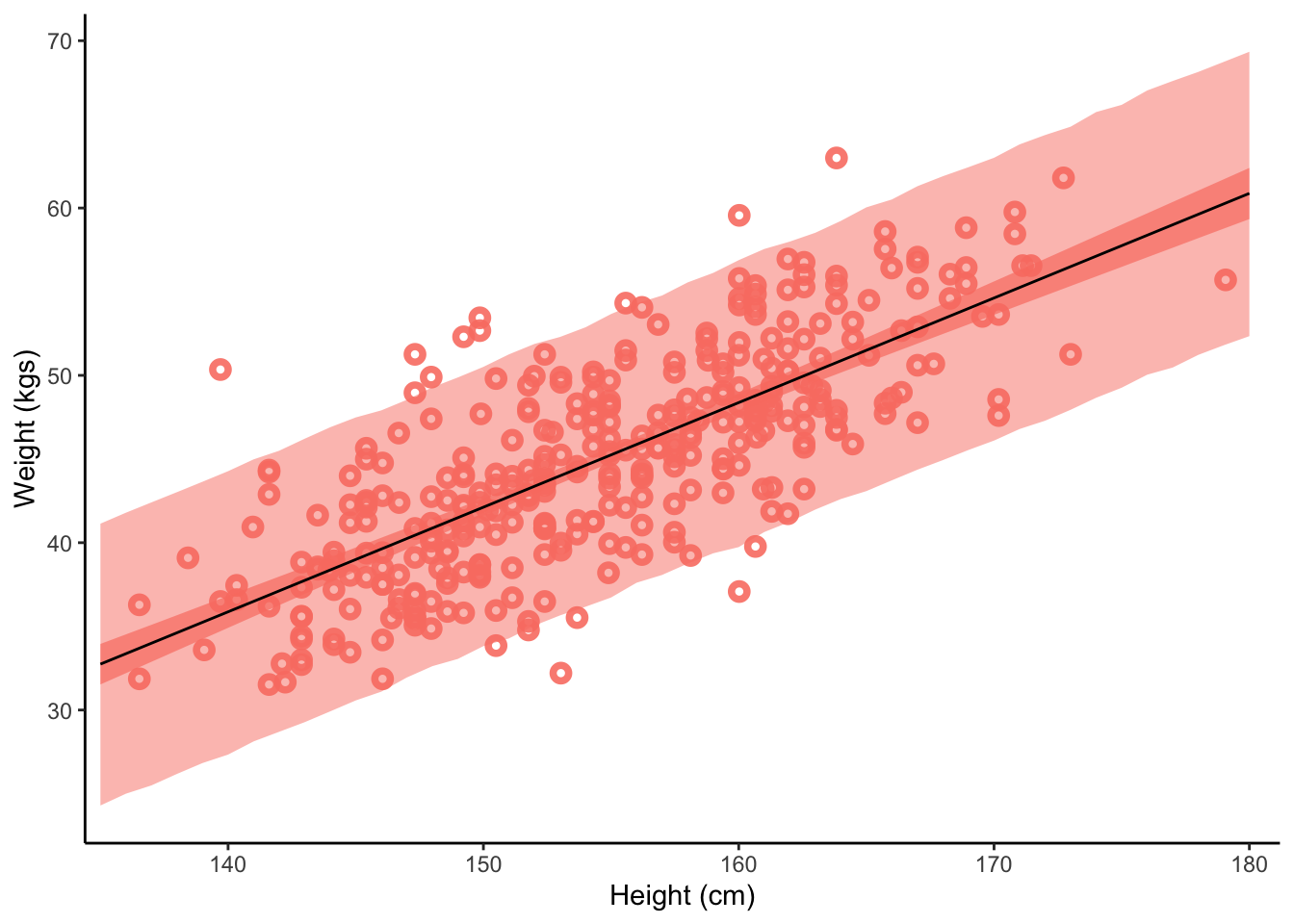
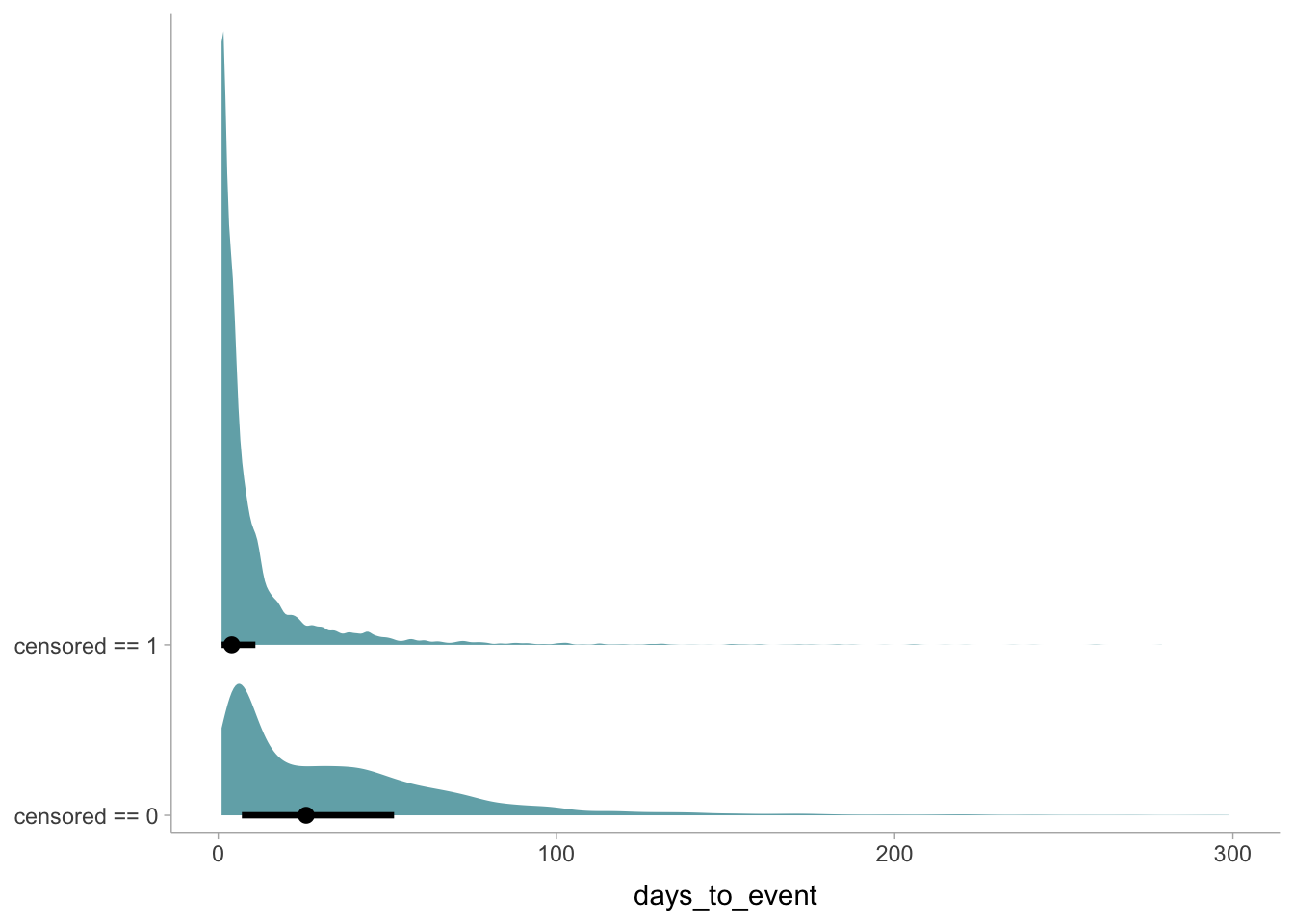
This plot shows 1) the raw data, 2) the predicted relationship between height and weight with 3) 95% uncertainty intervals for the mean and 4) 95% prediction intervals for where data points are predicted to fall within.
Predict() reports prediction intervals, which are simulations that are the joint consequence of both the mean and sigma, unlike the results of fitted(), which only reflect the mean.
\(~\)
Lecture 4: Categories and Curves
\(~\)
Categories
Want to stratify by categories in our data. This means fitting a separate regression line for each category.
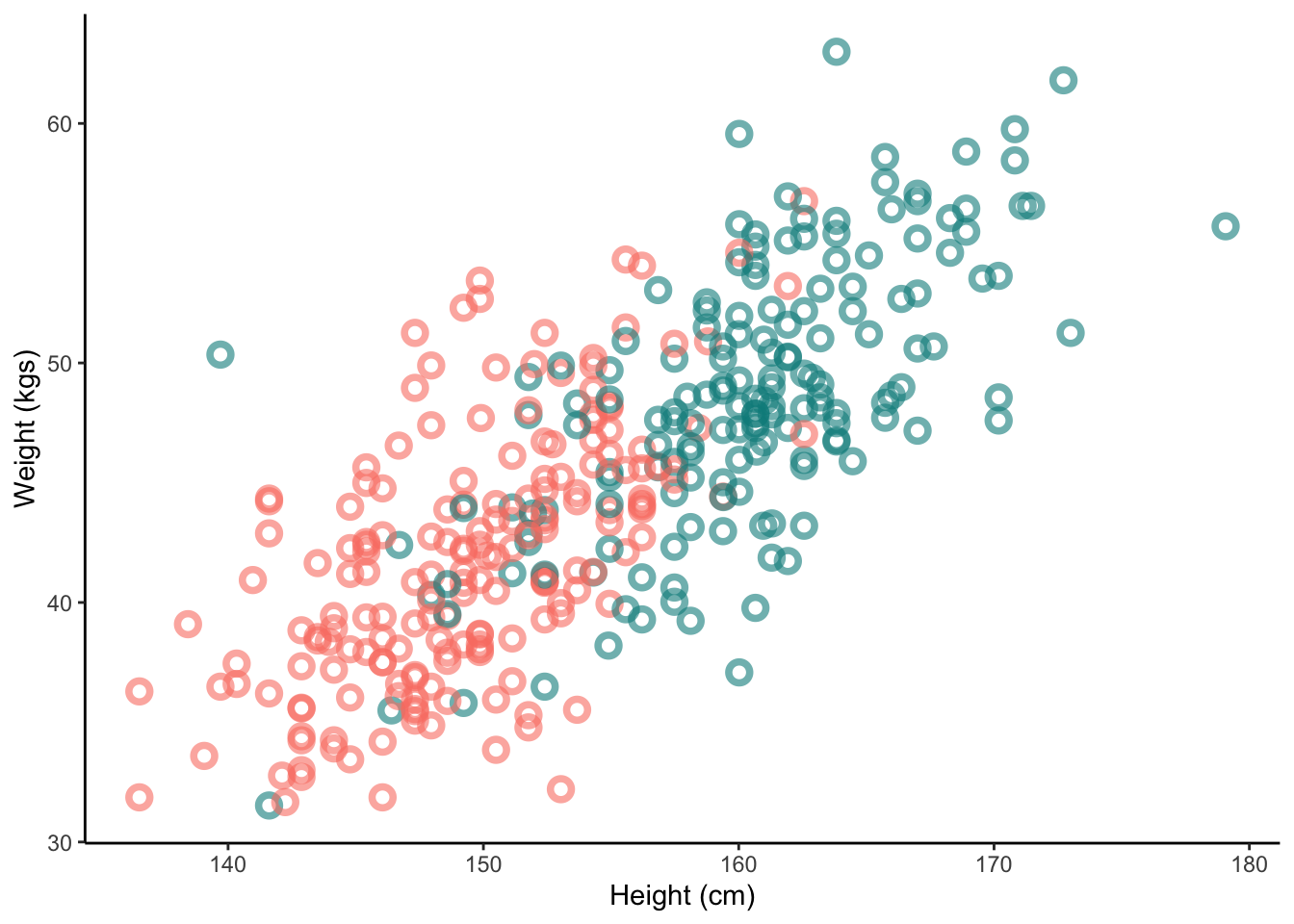
Lets return to the Kalahari data. Now we’ll add in a categorical variable - the sex of the individual.
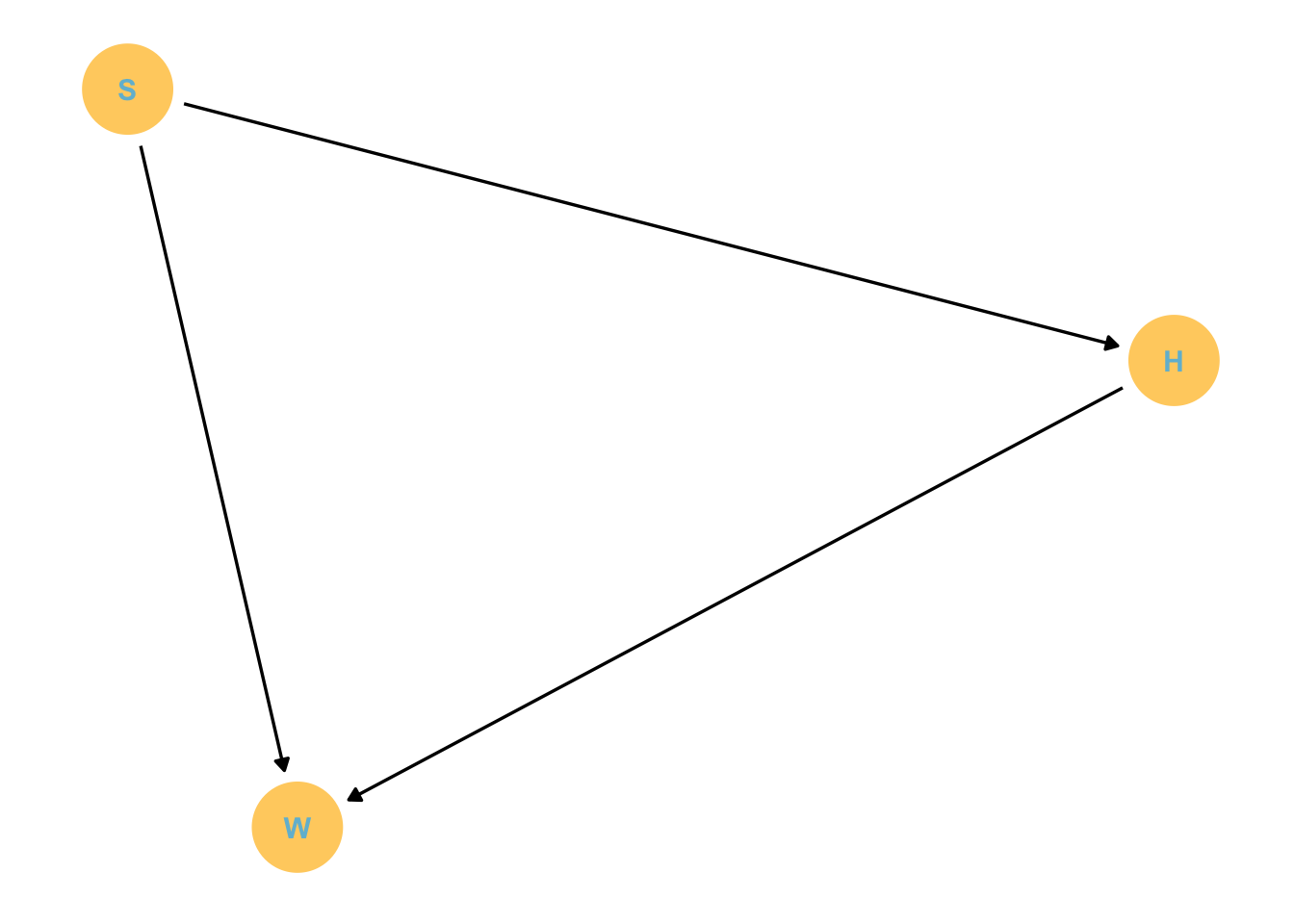
How are height, sex and weight causally associated?
How are height, sex and weight statistically related?
Lets build a DAG:
First, we know that height causally effects weight. You can change your own weight without changing your height. However, as you get taller there is more of you, thus it is reasonable to expect height to affect weight.
We also know that sex influences height, but it is silly to say that height influences sex.
Third, if there is any influence we expect sex to influence weight not the other way around. Therefore weight may be influenced by both height and sex.
Lets draw this DAG
Code
dagify(W ~ S + H, H ~ S,labels =c("W"="Weight", "H"="Height","S"="Sex")) %>%gg_simple_dag()
This is a mediation graph. Note that effects do not stop at one variable, instead they continue on through the path. In that way sex has both a direct and indirect effect on weight. The indirect effect may be through height, which is ‘contaminated’ by sex.
Statistically:
\(H = f_{H}(S)\)
\(W = f_{W}(H, S)\)
Following our workflow lets simulate a more complex dataset, that this time includes separate sexes.
To keep in line with the Kalahari data we’ll code females = 1 and males = 2
Code
sim_HW <-function(S, b, a){ N <-length(S) H <-if_else(S ==1, 150, 160) +rnorm(N, 0, 5) W <- a[S] + b[S]*H +rnorm(N, 0, 5)tibble(S, H, W)}
Give it some data and run sim_HW()
Code
S <- rethinking::rbern(100) +1(synthetic_data <-sim_HW(S, b =c(0.5, 0.6), a =c (0, 0)))
# A tibble: 100 × 3
S H W
<dbl> <dbl> <dbl>
1 2 161. 98.8
2 1 156. 81.0
3 1 148. 65.0
4 2 161. 98.2
5 1 148. 80.3
6 1 156. 75.2
7 1 142. 71.9
8 2 155. 94.0
9 2 162. 95.8
10 2 156. 95.1
# … with 90 more rows
Define the questions we’re going to ask:
Different questions lead to a need for different stats models.
Q: Causal effect of H on W?
Q: Causal effect of S on W?
Q: Direct effect of S on W?
Each require different components of the DAG.
Drawing the categorical OWL:
There are several ways to code categorical variables
Dummy or indicator variables
Series of 0 1 variables that stand in for categories
Index variables
Assign an index value to each category
Better for specifying priors
Extend effortlessly to multi-level models
What we will use
\(~\)
Q: What is the causal effect of S on W?
\(~\)
Using index variables
Estimating average weight:
\(W_{i} = Normal(\mu_{i}, \sigma)\)
\(\mu_{i} = \alpha S[i]\) where \(S[i]\) is the sex of the i-th person
S = 1 indicates female, S = 2 indicates male
\(\alpha = [\alpha_{i}, \alpha_{2}]\) this means there are two intercepts, one for each sex
Priors
\(\alpha_{j} = Normal(60, 10)\)
All this says is that there is more than one value of (indicated by the subscript j) and that we want the prior to be the same for each of the values. Values correspond to the categories.
We’ll let the sample update the prior and tell us if sexual dimorphism exists
Right ready to model. Not yet! More simulation. This might seem like overkill but it will help so much to get into this habit.
We’ll construct a female and male sample and look at the average difference. We’ll only change sex.
We’ll find the difference between the sexes in our simulation (remember we coded a stronger effect of height on male weight than on female weight):
Code
# female sampleS <-rep(1, 100)simF <-sim_HW(S, b =c(0.5, 0.6), a =c(0, 0))S <-rep(2, 100)simM <-sim_HW(S, b =c(0.5, 0.6), a =c(0, 0))# effect of Sex (male - female)mean(simM$W - simF$W)
[1] 19.53385
Ok a difference of ~21kgs
Now lets test the estimator.
Note that we have specified a 0 intercept. In brms this will result in an output that produces a separate intercept for each categorical variable.
Code
synthetic_kalahari_sex_model <-brm(data = synthetic_data, W ~0+as.factor(S),prior =c(prior(normal(60, 10), class = b),prior(exponential(1), class = sigma)),iter =2000, warmup =1000, chains =4, cores =4,seed =1,file ="fits/synthetic_kalahari_sex")synthetic_kalahari_sex_model
Family: gaussian
Links: mu = identity; sigma = identity
Formula: W ~ 0 + as.factor(S)
Data: synthetic_data (Number of observations: 100)
Draws: 4 chains, each with iter = 2000; warmup = 1000; thin = 1;
total post-warmup draws = 4000
Population-Level Effects:
Estimate Est.Error l-95% CI u-95% CI Rhat Bulk_ESS Tail_ESS
as.factorS1 75.69 0.82 74.13 77.31 1.00 4038 2899
as.factorS2 96.13 0.74 94.69 97.63 1.00 4126 2946
Family Specific Parameters:
Estimate Est.Error l-95% CI u-95% CI Rhat Bulk_ESS Tail_ESS
sigma 5.50 0.36 4.86 6.29 1.00 4199 2984
Draws were sampled using sampling(NUTS). For each parameter, Bulk_ESS
and Tail_ESS are effective sample size measures, and Rhat is the potential
scale reduction factor on split chains (at convergence, Rhat = 1).
Again we find a difference between the sexes ~21kgs. Looks like our model tests what we want to test.
Now lets use the real data
Code
# make the index variable to match McElreath's datasetKalahari_adults <- Kalahari_adults %>%mutate(Sex =if_else(male =="1", 2, 1),Sex =as.factor(Sex))kalahari_sex_model <-brm(data = Kalahari_adults, weight ~0+ Sex,prior =c(prior(normal(60, 10), class = b),prior(exponential(1), class = sigma)),iter =2000, warmup =1000, chains =4, cores =4,seed =1,file ="fits/_kalahari_sex_model")
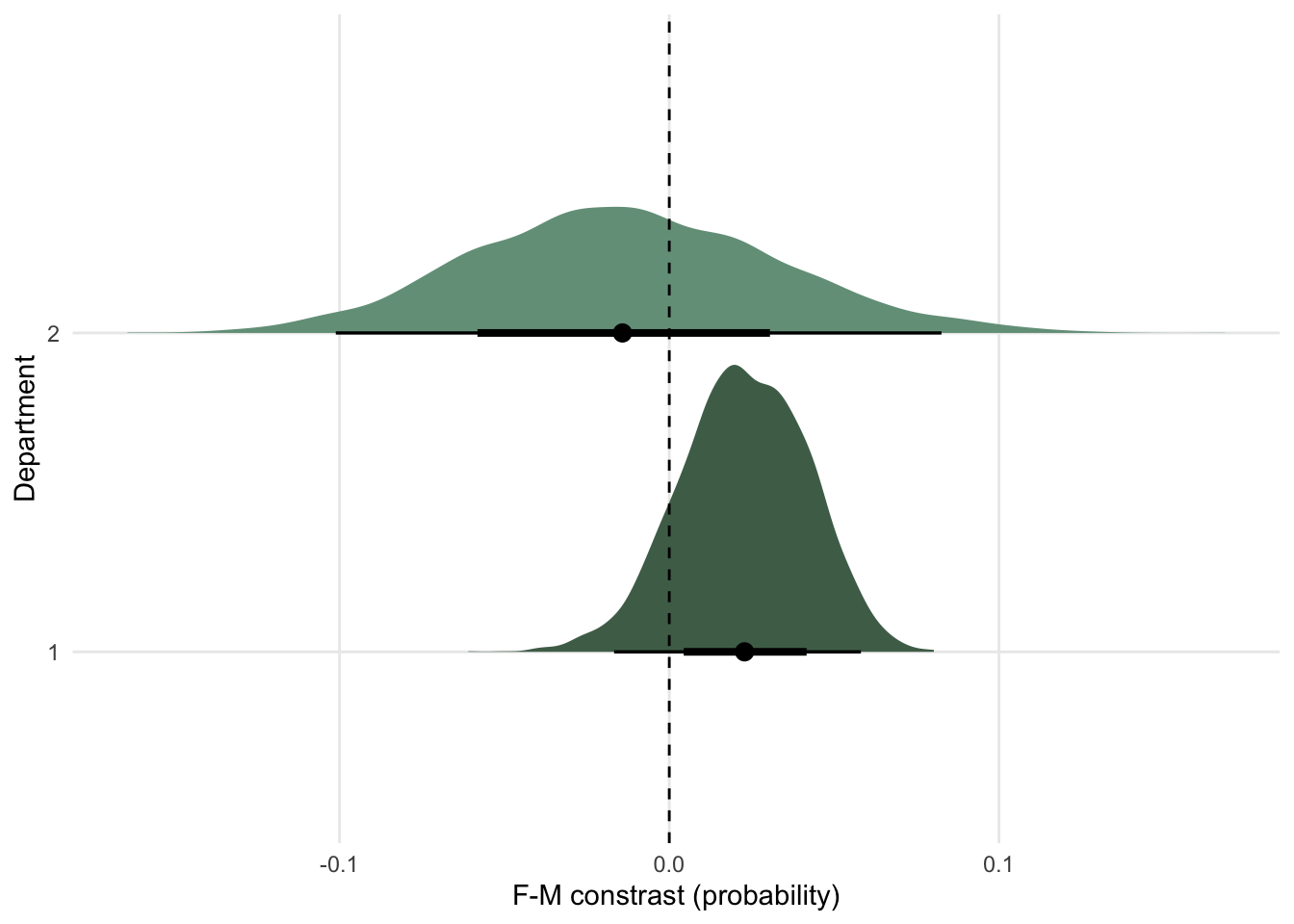
Find the contrasts and plot the average differences between women and men
Kalahari_h_S_model <-brm(data = Kalahari_adults, weight ~0+ Sex + height_standard,prior =c(#prior(normal(60, 10), class = a),prior(lognormal(0, 1), class = b, lb =0),prior(exponential(1), class = sigma)),iter =4000, warmup =2000, chains =4, cores =4,seed =1,file ="fits/Kalahari_h_S_model")
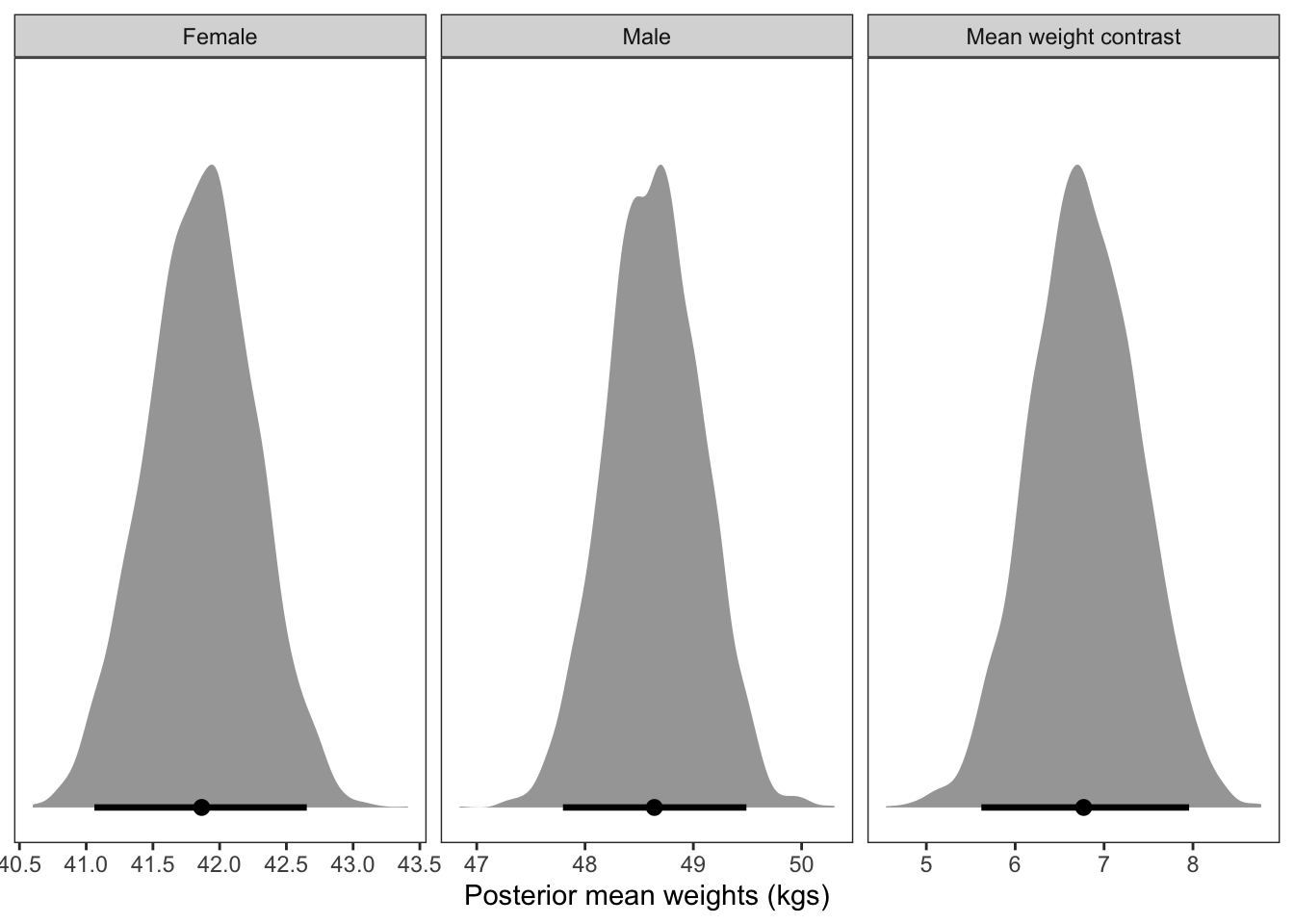
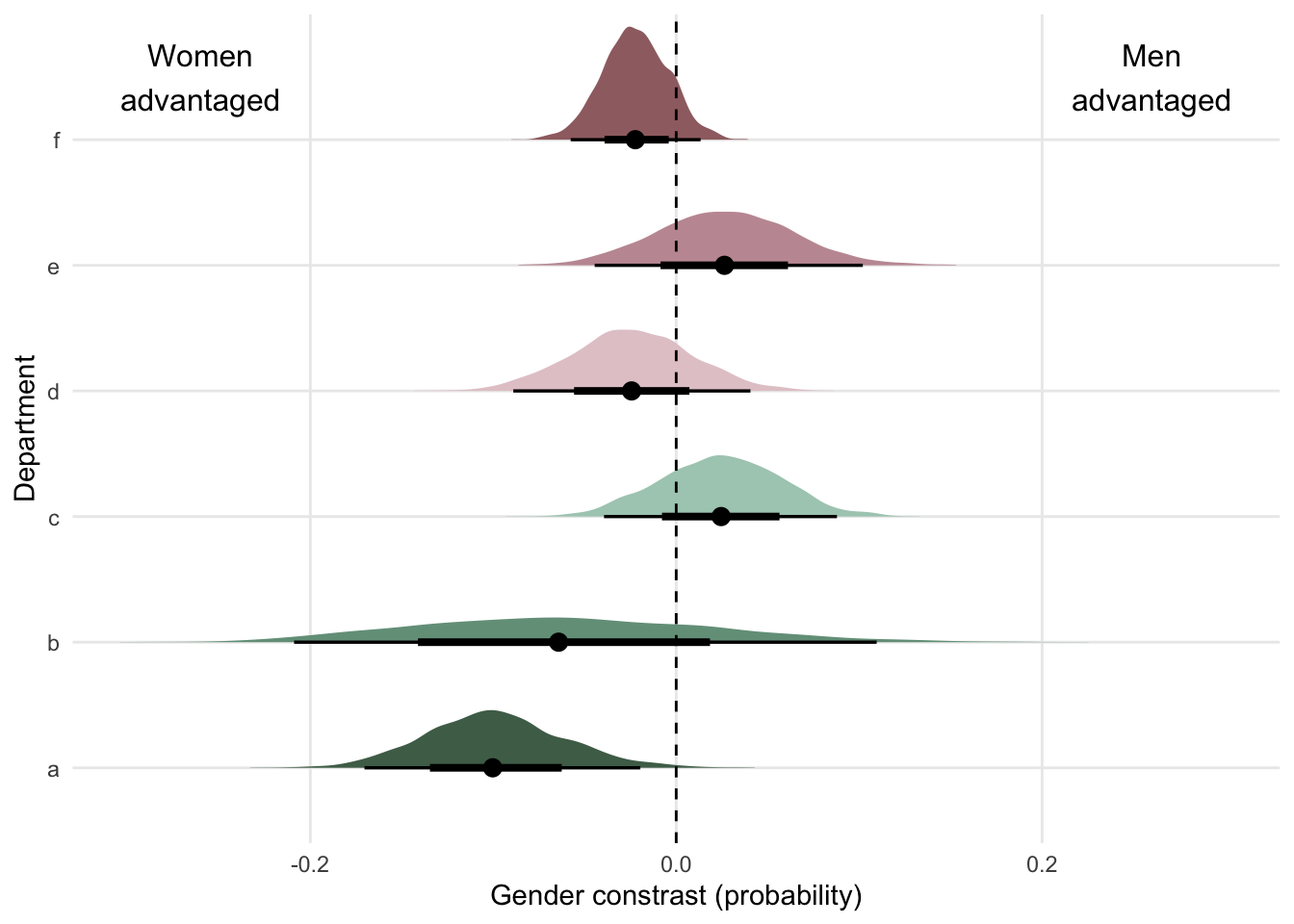
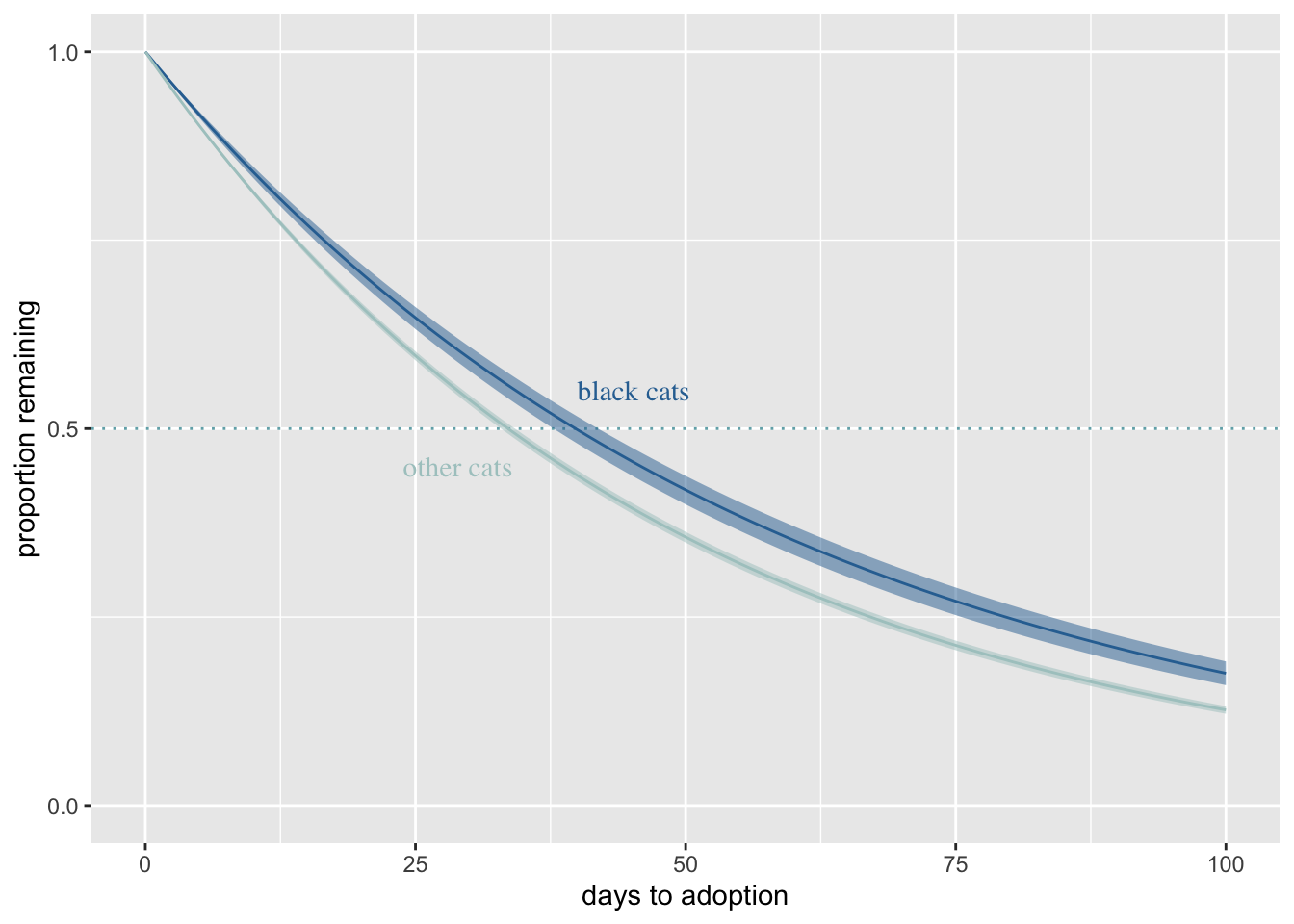
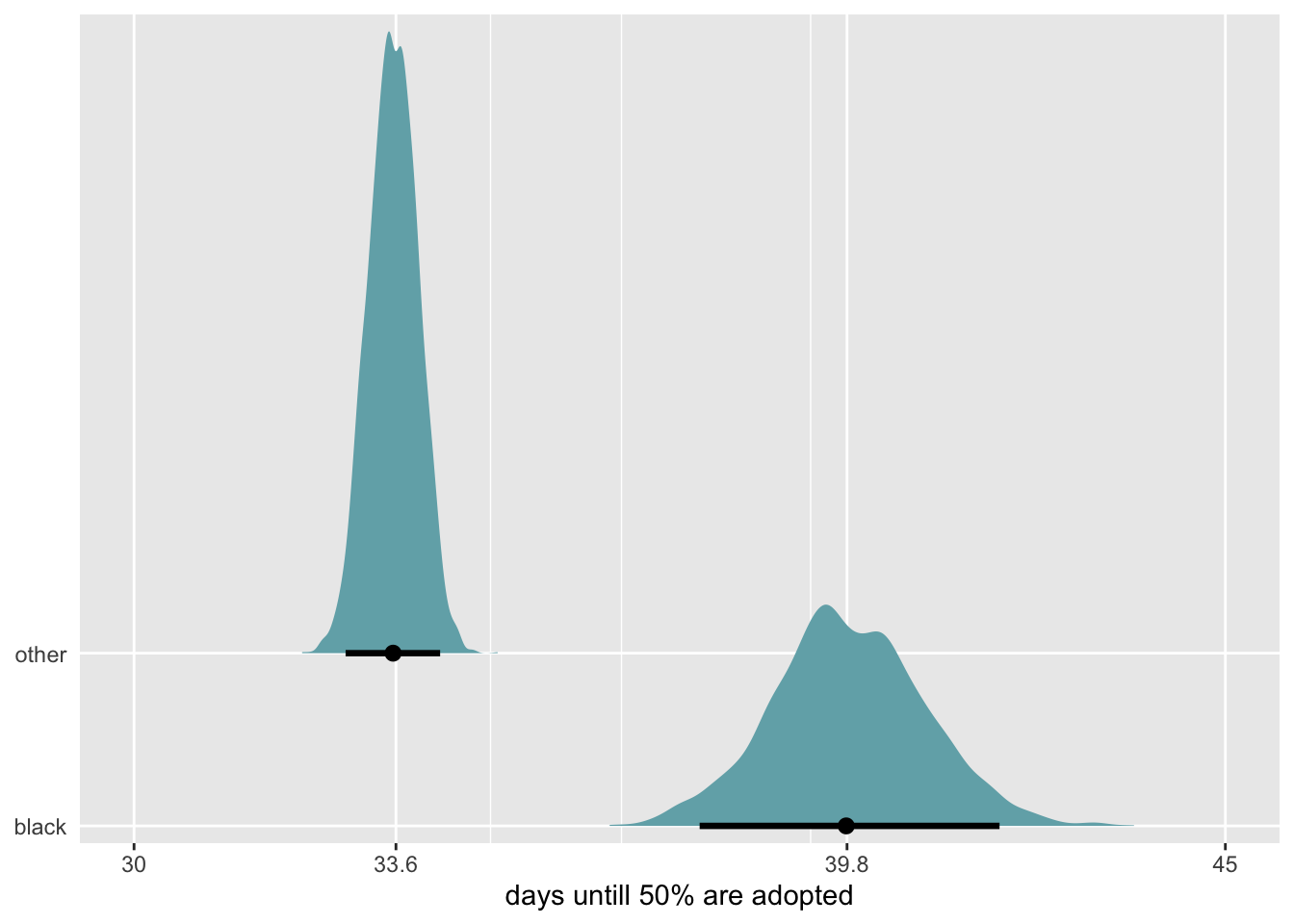
I’m not sure how to make McElreath’s plot here - he brilliantly finds the effect of sex at 50 different heights then plots. The best I can do is the overall effect of sex on weight after accounting for sex.
The takeaway however, is that nearly all of the causal effect of sex acts through height.
Code
draws_2 <-as_draws_df(Kalahari_h_S_model) %>%mutate(weight_contrast = b_Sex2 - b_Sex1)Female_dist_2 <-rnorm(1000, draws_2$b_Sex1, draws_2$sigma) %>%as_tibble() %>%rename(Female = value)Male_dist_2 <-rnorm(1000, draws_2$b_Sex2, draws_2$sigma) %>%as_tibble() %>%rename(Male = value)plot_1 <-cbind(Female_dist_2, Male_dist_2) %>%pivot_longer(cols =c("Female", "Male")) %>%ggplot(aes(x = value, group = name, colour = name, fill = name)) +geom_density(alpha =0.8) +scale_colour_manual(values =c("salmon", "darkcyan")) +scale_fill_manual(values =c("salmon", "darkcyan")) +xlab("Posterior predicted weights (kgs)\nafter accounting for the effect of height") +ylab("Density") +theme_bw() +theme(panel.grid =element_blank(),legend.position ="none")plot_2 <-cbind(Female_dist, Male_dist) %>%as_tibble() %>%mutate(predictive_diff = Male - Female) %>%ggplot(aes(x = predictive_diff, colour ="orange", fill ="orange")) +geom_density(alpha =0.8) +xlab("Posterior predictive weight difference (kgs)\nafter accounting for the effect of height") +ylab("Density") +theme_bw() +theme(panel.grid =element_blank(),legend.position ="none")plot_1 / plot_2
\(~\)
Curves with lines
\(~\)
Many causal relationships are obviously not linear.
Linear models can fit curves quite easily, but be wary that this is geocentric (not mechanistic).
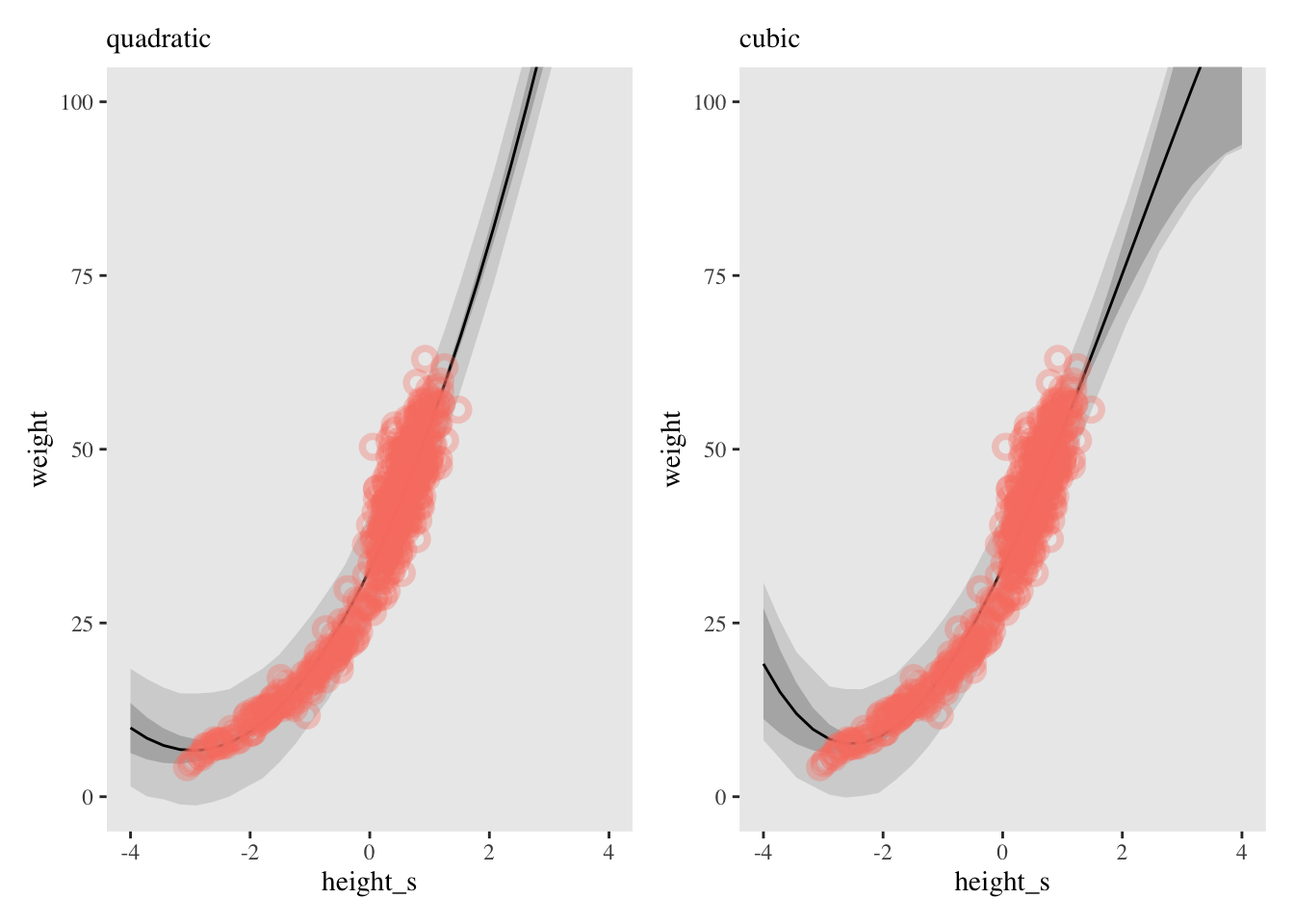
For example lets examine the full Kalahari dataset, which includes children.
Note the \(x^{2}\), you can use higher and higher order terms and the model becomes more flexible.
Issues
There is unhelpful asyymetry to polynomials. For example, for a second order term, the model believes the data creates a parabola and it’s very hard to convince it otherwise.
The uncertainty at the edges of the data can be huge, making extrapolation and prediction very difficult.
There is no local smoothing so any data point can massively change the shape of a curve.
For a second order term, the model believes the data creates a parabola and it’s very hard to convince it otherwise.
Note our use of the coef argument within our prior statements. Since \(\beta_{1}\) and \(\beta_{2}\) are both parameters of class = b within the brms set-up, we need to use the coef argument in cases when we want their priors to differ.
At the left extreme, the model predicts that weight will increase as height decreases. This is because we have told the model the data is curved.
\(~\)
Splines
\(~\)
A big family of functions designed to do local smoothing.
This means that only the points within a region determine the shape of the function in that region. In polynomials the whole shape is affected by each data point. Results in splines being far more flexible than polynomials.
Once again, they have no mechanistic reality.
What is a spline?
Used in drafting for architecture. They create wriggly lines.
\(w\) = the weight parameter - like a slope, determine the importance of the different variables for the predicted mean. These can overlap somewhat.
\(B\) = the basis function - you make this - only have positive values in narrow regions of x-axis. They turn on weights in isolated regions of the x-axis.
\(~\)
Lecture 5: Elemental Confounds
\(~\)
Correlation in general is not surprising. In large data sets, every pair of variables has a statistically discernible non-zero correlation. But since most correlations do not indicate causal relationships, we need tools for distinguishing mere association from evidence of causation. This is why so much effort is devoted to multiple regression, using more than one predictor variable to simultaneously model an outcome.
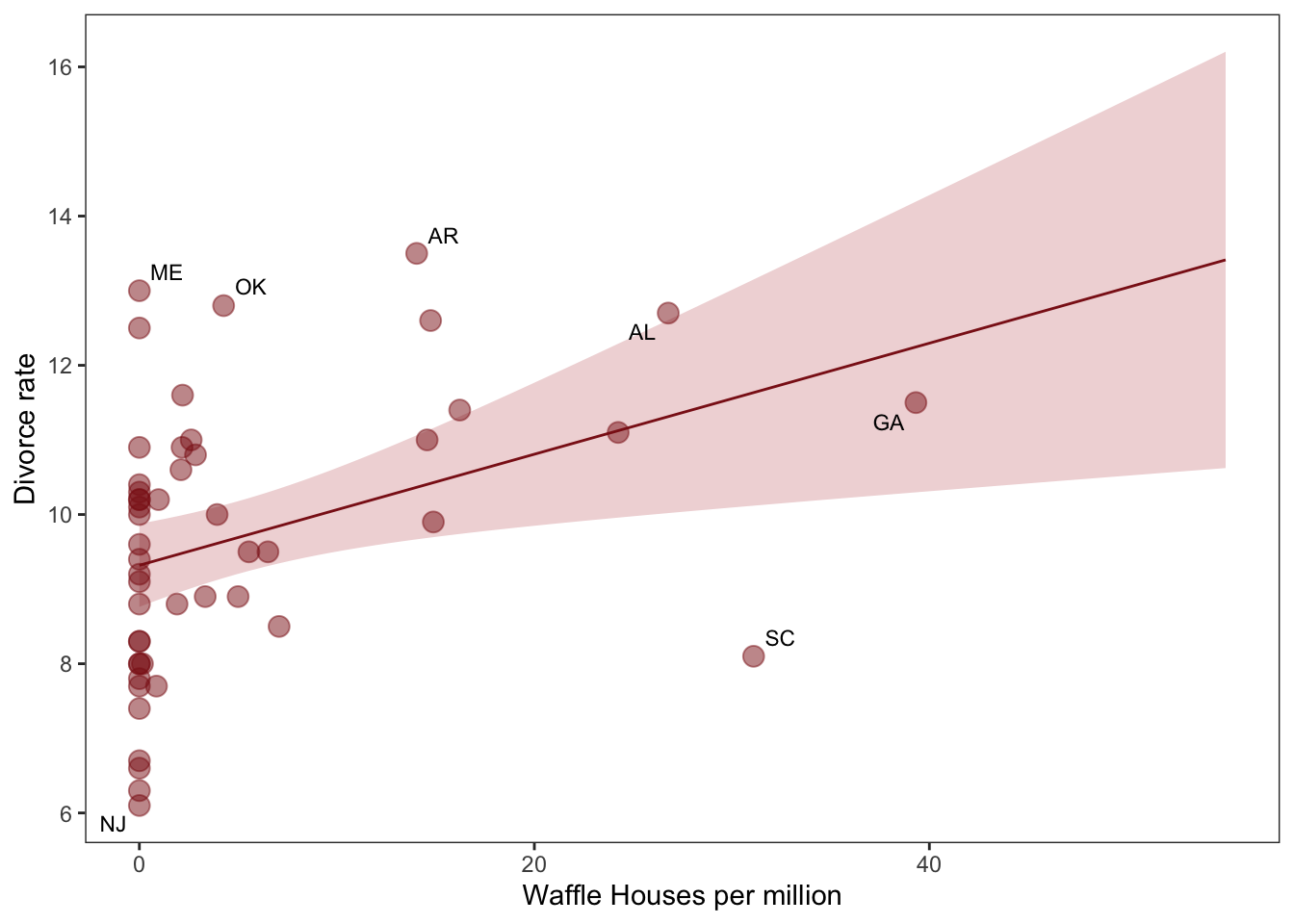
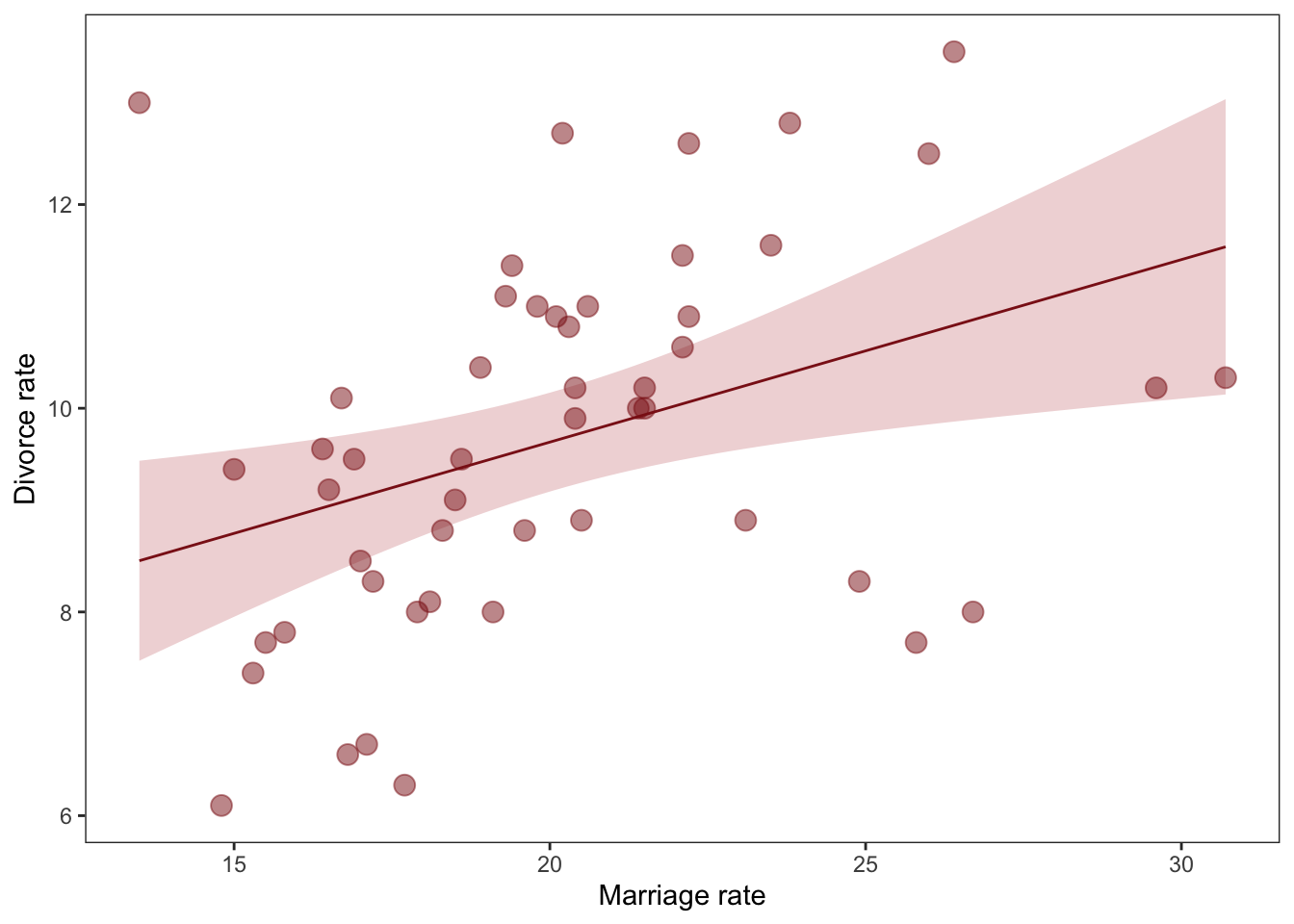
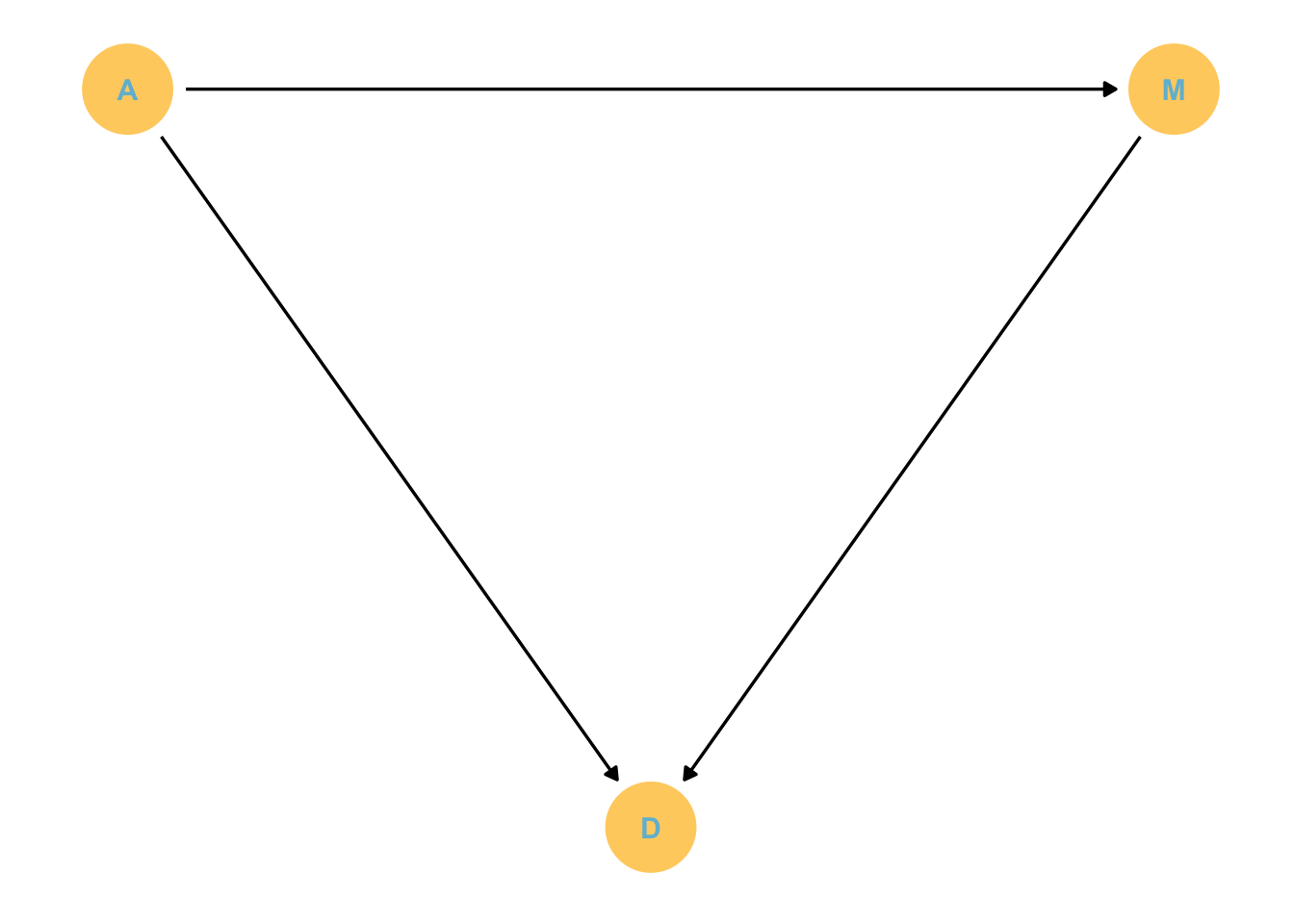
Waffle house causes divorce?
Code
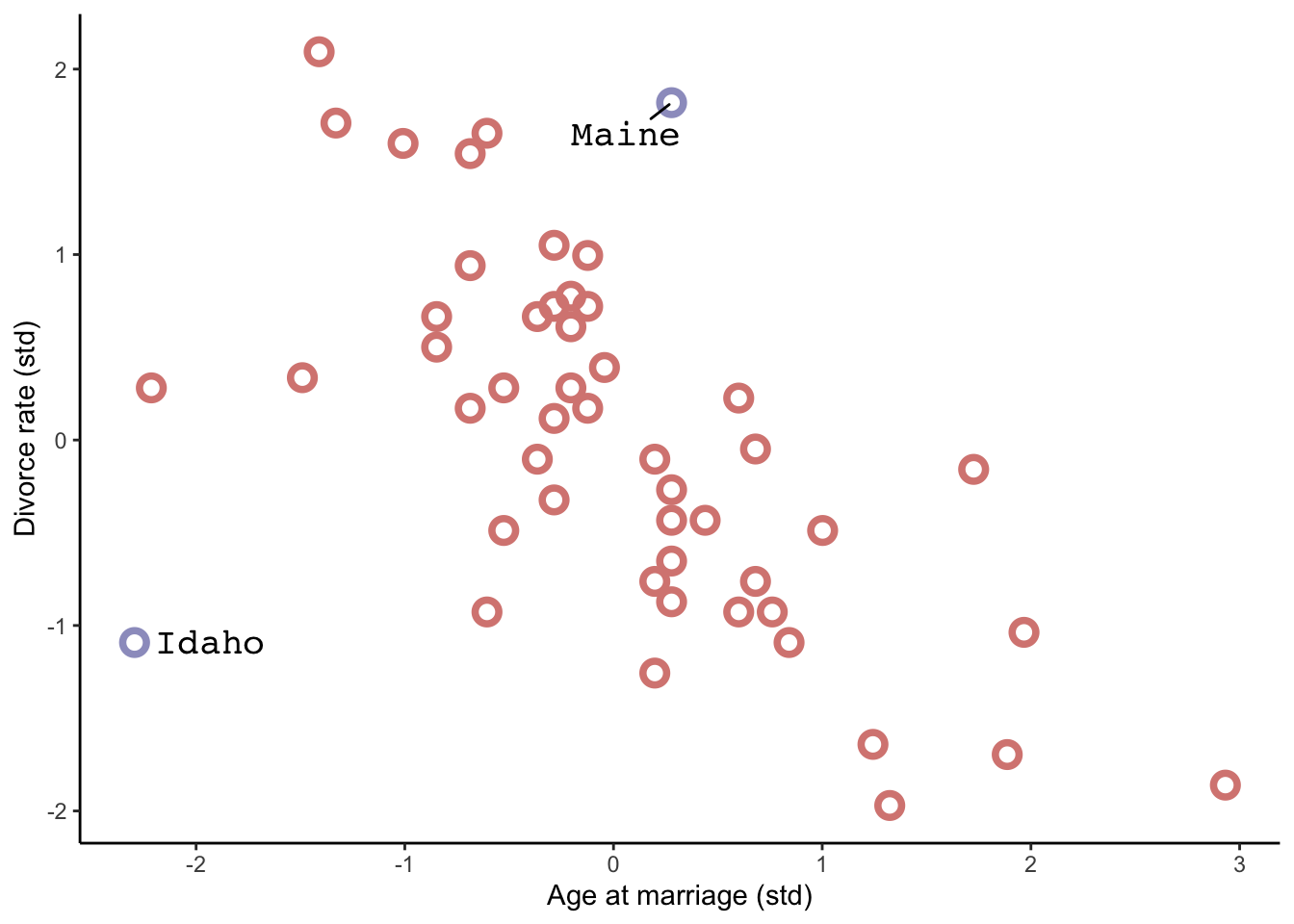
data(WaffleDivorce, package ="rethinking") # this loads WaffleDivorce as a dataframe without loading rehtinking# Standarise as discussed below. We can use the rethinking packageWaffle_data <- WaffleDivorce %>%mutate(d = rethinking::standardize(Divorce),m = rethinking::standardize(Marriage),a = rethinking::standardize(MedianAgeMarriage))# time to plotWaffle_data %>%ggplot(aes(x = WaffleHouses/Population, y = Divorce)) +stat_smooth(method ="lm", fullrange = T, size =1/2,color ="firebrick4", fill ="firebrick", alpha =1/5) +geom_point(size =3.5, color ="firebrick4", alpha =1/2) +geom_text_repel(data = Waffle_data %>%filter(Loc %in%c("ME", "OK", "AR", "AL", "GA", "SC", "NJ")), aes(label = Loc), size =3, seed =1042) +# this makes it reproduciblescale_x_continuous("Waffle Houses per million", limits =c(0, 55)) +ylab("Divorce rate") +theme_bw() +theme(panel.grid =element_blank())
There is a statistical association! But surely this isn’t casual. We’ll get back to this later.
\(~\)
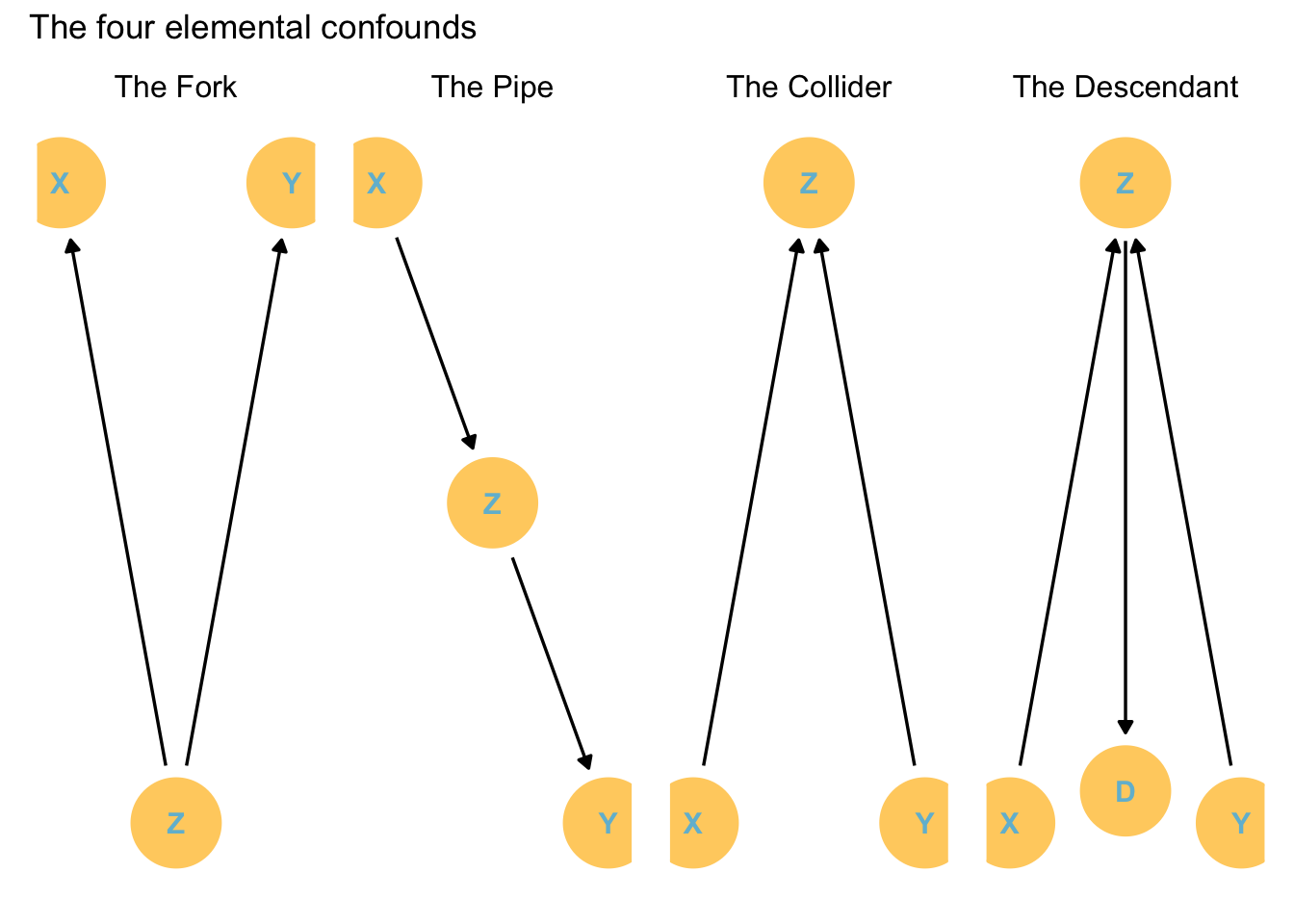
The four elemental confounds
\(~\)
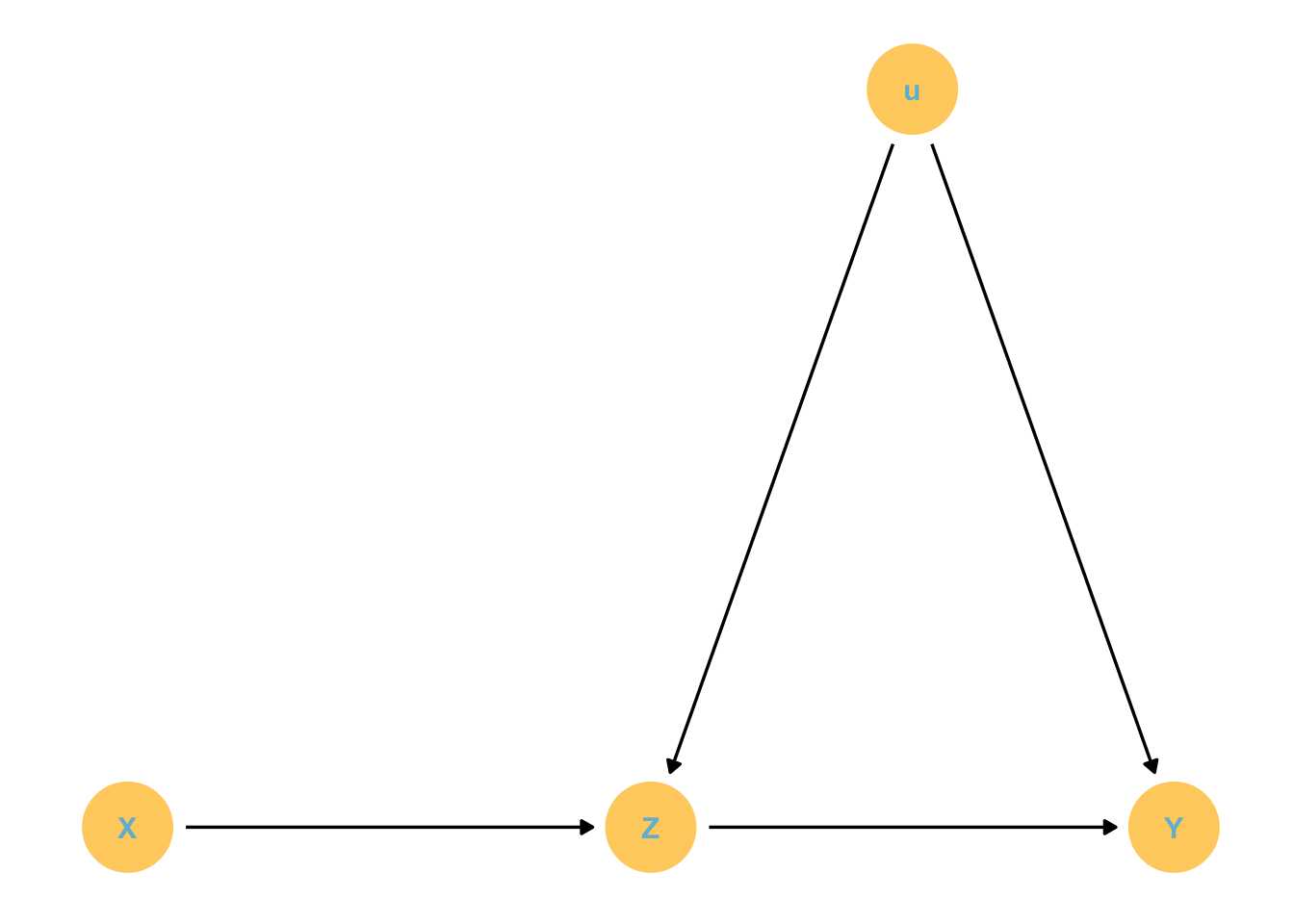
Confounds: some feature of the sample and how we use it that misleads us.
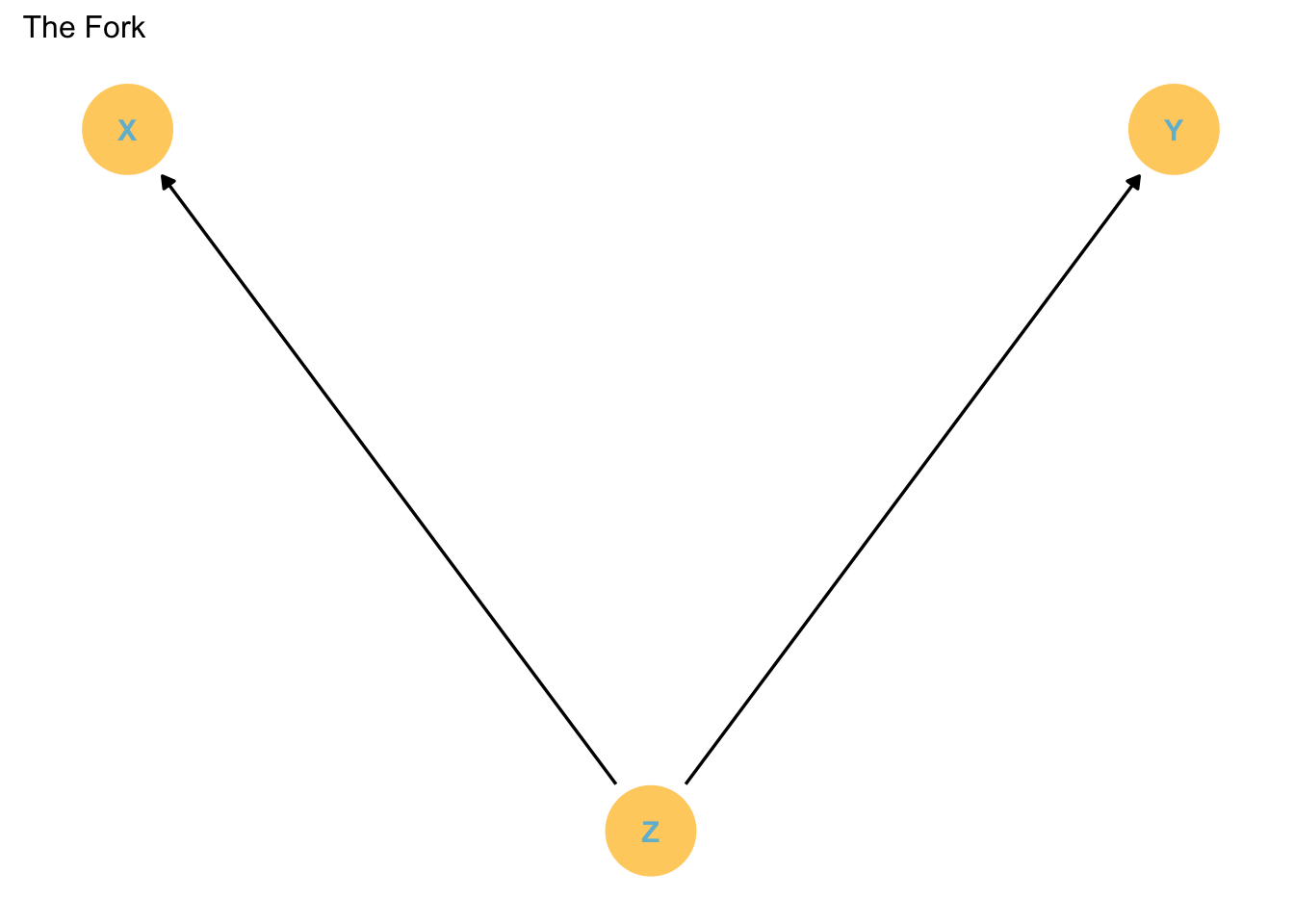
X and Y are associated because they share a common cause. However, this association disappears once we account for Z. That means that Y is independent of X once we account for Z.
Code
p1
Note that once we stratify for Z, the noise affecting X and Y can become independent.
Let’s simulate:
Code
n <-1000tibble(Z =rbernoulli(n, 0.5)) %>%mutate(Z =if_else(Z =="TRUE", 1, 0),X =rbernoulli(n, (1- Z) *0.1+ Z *0.9),Y =rbernoulli(n, (1- Z)*0.1+ Z *0.9),X =if_else(X =="TRUE", 1, 0),Y =if_else(Y =="TRUE", 1, 0)) %>%count(X, Y)
# A tibble: 4 × 3
X Y n
<dbl> <dbl> <int>
1 0 0 425
2 0 1 97
3 1 0 77
4 1 1 401
See how this works? Our simulation specifies no relation between X and Y except through Z, but they are super positively correlated.
Before we talk about the exponential prior lets standardise the data (done above but explained here)
Why standardise?
To standardise subtract the mean and divide each value by the standard deviation. Variables become Z scores - values represent SDs from the mean. 0 is the mean.
This makes computation more efficient.
Priors are easier to choose because we have some understanding about effect size.
Lets do some prior simulation, first with weak priors:
\(\alpha = Normal(0, 10)\)
\(\beta_{M} = Normal(0, 10)\)
\(\beta_{A} = Normal(0, 10)\)
\(\sigma = Exponential(1)\)
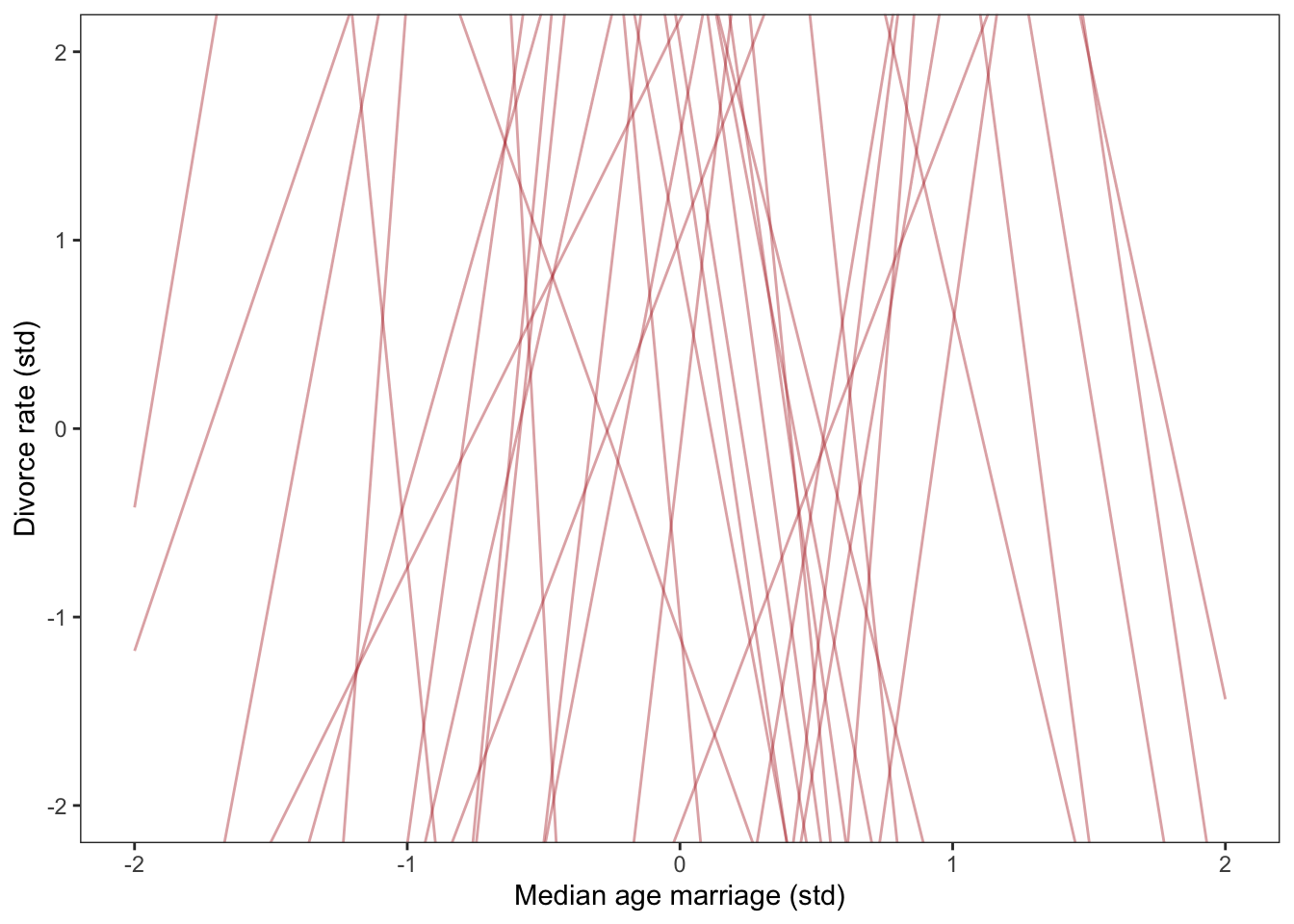
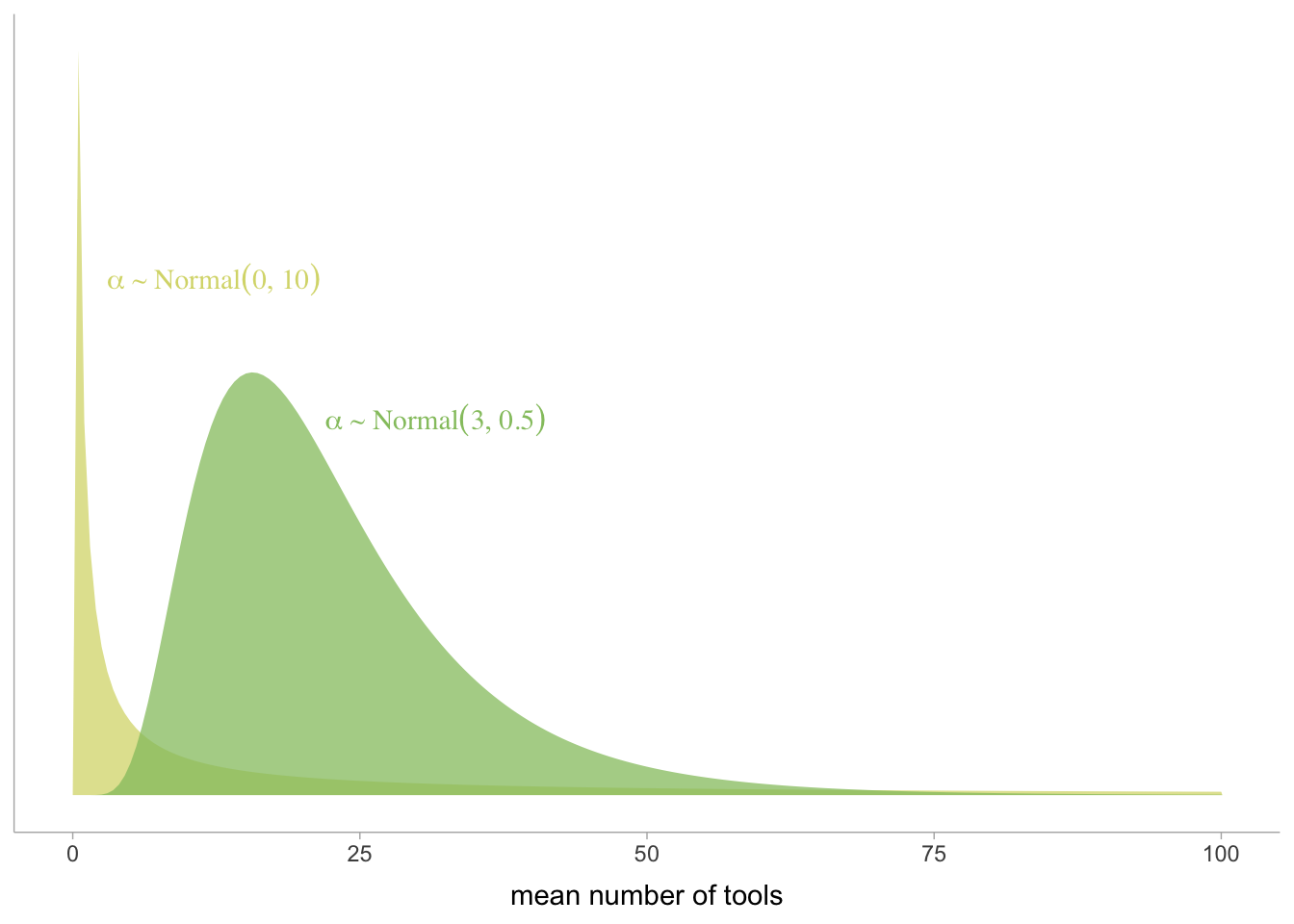
These priors are so broad they are essentially flat. This is a bad idea. The reason is because almost all the probability mass is for insanely strong pos or neg relationships.
The plot below shows the stupidly steep slopes these priors predict.
Code
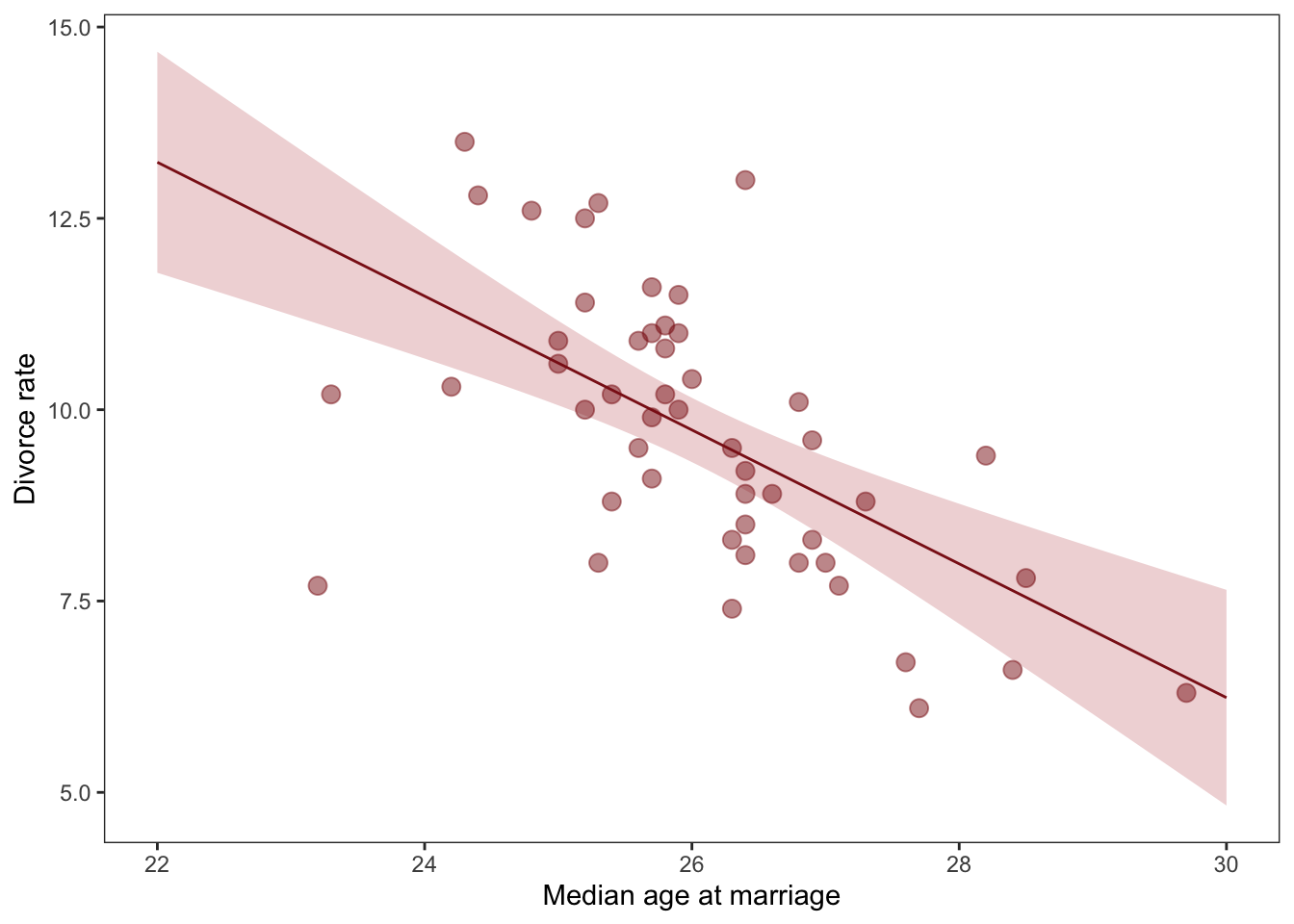
# fit a model with bad priorsWaffle_model_bad_priors <-brm(data = Waffle_data, family = gaussian, d ~1+ a,prior =c(prior(normal(0, 10), class = Intercept),prior(normal(0, 10), class = b),prior(exponential(1), class = sigma)),iter =2000, warmup =1000, chains =4, cores =4,seed =5,sample_prior = T,file ="fits/Waffle_model_bad_priors")set.seed(5)prior_draws(Waffle_model_bad_priors) %>%slice_sample(n =50) %>%rownames_to_column("draw") %>%expand(nesting(draw, Intercept, b),a =c(-2, 2)) %>%mutate(d = Intercept + b * a) %>%ggplot(aes(x = a, y = d)) +geom_line(aes(group = draw),color ="firebrick", alpha = .4) +labs(x ="Median age marriage (std)",y ="Divorce rate (std)") +coord_cartesian(ylim =c(-2, 2)) +theme_bw() +theme(panel.grid =element_blank())
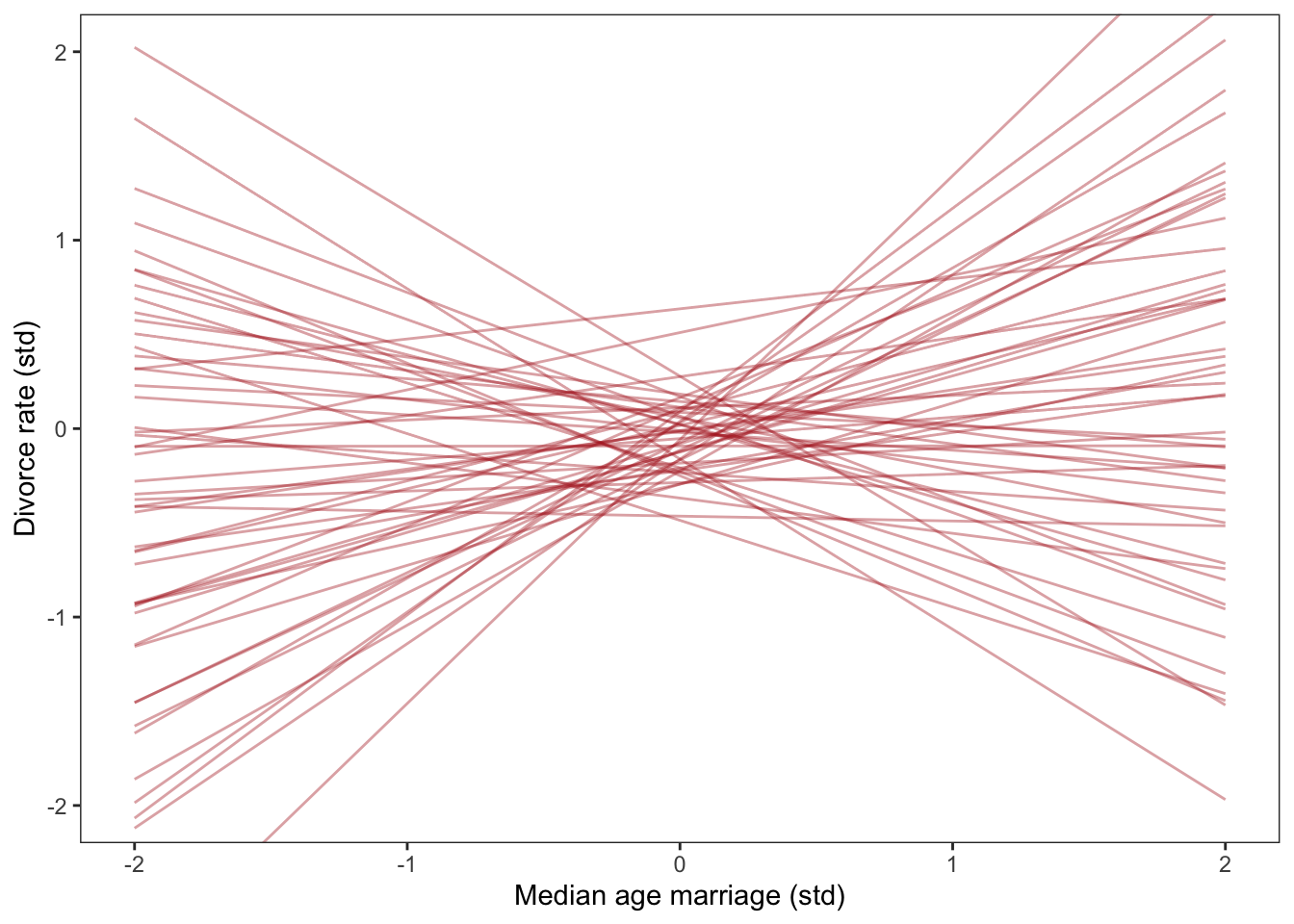
Better priors
\(\alpha = Normal(0, 0.2)\)
\(\beta_{M} = Normal(0, 0.5)\)
\(\beta_{A} = Normal(0, 0.5)\)
\(\sigma = Exponential(1)\)
Re-sample
Code
Marriage_age_model <-brm(data = Waffle_data, family = gaussian, d ~1+ a,prior =c(prior(normal(0, 0.2), class = Intercept),prior(normal(0, 0.5), class = b),prior(exponential(1), class = sigma)),iter =2000, warmup =1000, chains =4, cores =4,seed =5,sample_prior = T,file ="fits/Marriage_age_model")Marriage_age_model
Family: gaussian
Links: mu = identity; sigma = identity
Formula: d ~ 1 + a
Data: Waffle_data (Number of observations: 50)
Draws: 4 chains, each with iter = 2000; warmup = 1000; thin = 1;
total post-warmup draws = 4000
Population-Level Effects:
Estimate Est.Error l-95% CI u-95% CI Rhat Bulk_ESS Tail_ESS
Intercept 0.00 0.10 -0.20 0.20 1.00 3537 2678
a -0.57 0.12 -0.79 -0.34 1.00 3801 2753
Family Specific Parameters:
Estimate Est.Error l-95% CI u-95% CI Rhat Bulk_ESS Tail_ESS
sigma 0.82 0.09 0.68 1.01 1.00 2735 2675
Draws were sampled using sampling(NUTS). For each parameter, Bulk_ESS
and Tail_ESS are effective sample size measures, and Rhat is the potential
scale reduction factor on split chains (at convergence, Rhat = 1).
Lets have a look at that plot again with our updated priors
Code
prior_draws(Marriage_age_model) %>%slice_sample(n =50) %>%rownames_to_column("draw") %>%expand(nesting(draw, Intercept, b),a =c(-2, 2)) %>%mutate(d = Intercept + b * a) %>%ggplot(aes(x = a, y = d)) +geom_line(aes(group = draw),color ="firebrick", alpha = .4) +labs(x ="Median age marriage (std)",y ="Divorce rate (std)") +coord_cartesian(ylim =c(-2, 2)) +theme_bw() +theme(panel.grid =element_blank())
Much better!
Lets test for the causal effect of marriage, while closing the fork
Code
marriage_model <-brm(data = Waffle_data, family = gaussian, d ~1+ m + a,prior =c(prior(normal(0, 0.2), class = Intercept),prior(normal(0, 0.5), class = b),prior(exponential(1), class = sigma)),iter =2000, warmup =1000, chains =4, cores =4,seed =5,file ="fits/marriage_model")marriage_model
Family: gaussian
Links: mu = identity; sigma = identity
Formula: d ~ 1 + m + a
Data: Waffle_data (Number of observations: 50)
Draws: 4 chains, each with iter = 2000; warmup = 1000; thin = 1;
total post-warmup draws = 4000
Population-Level Effects:
Estimate Est.Error l-95% CI u-95% CI Rhat Bulk_ESS Tail_ESS
Intercept -0.00 0.10 -0.20 0.20 1.00 3514 2688
m -0.06 0.16 -0.38 0.26 1.00 2519 2660
a -0.61 0.16 -0.93 -0.28 1.00 2626 2584
Family Specific Parameters:
Estimate Est.Error l-95% CI u-95% CI Rhat Bulk_ESS Tail_ESS
sigma 0.83 0.09 0.68 1.02 1.00 3296 2324
Draws were sampled using sampling(NUTS). For each parameter, Bulk_ESS
and Tail_ESS are effective sample size measures, and Rhat is the potential
scale reduction factor on split chains (at convergence, Rhat = 1).
Ok the exponential distribution:
Constrained to be positive
Only info is average displacement from 0, hence the one value, also called the rate.
A value of 1 means roughly 1 SD from 0
Great for sigma
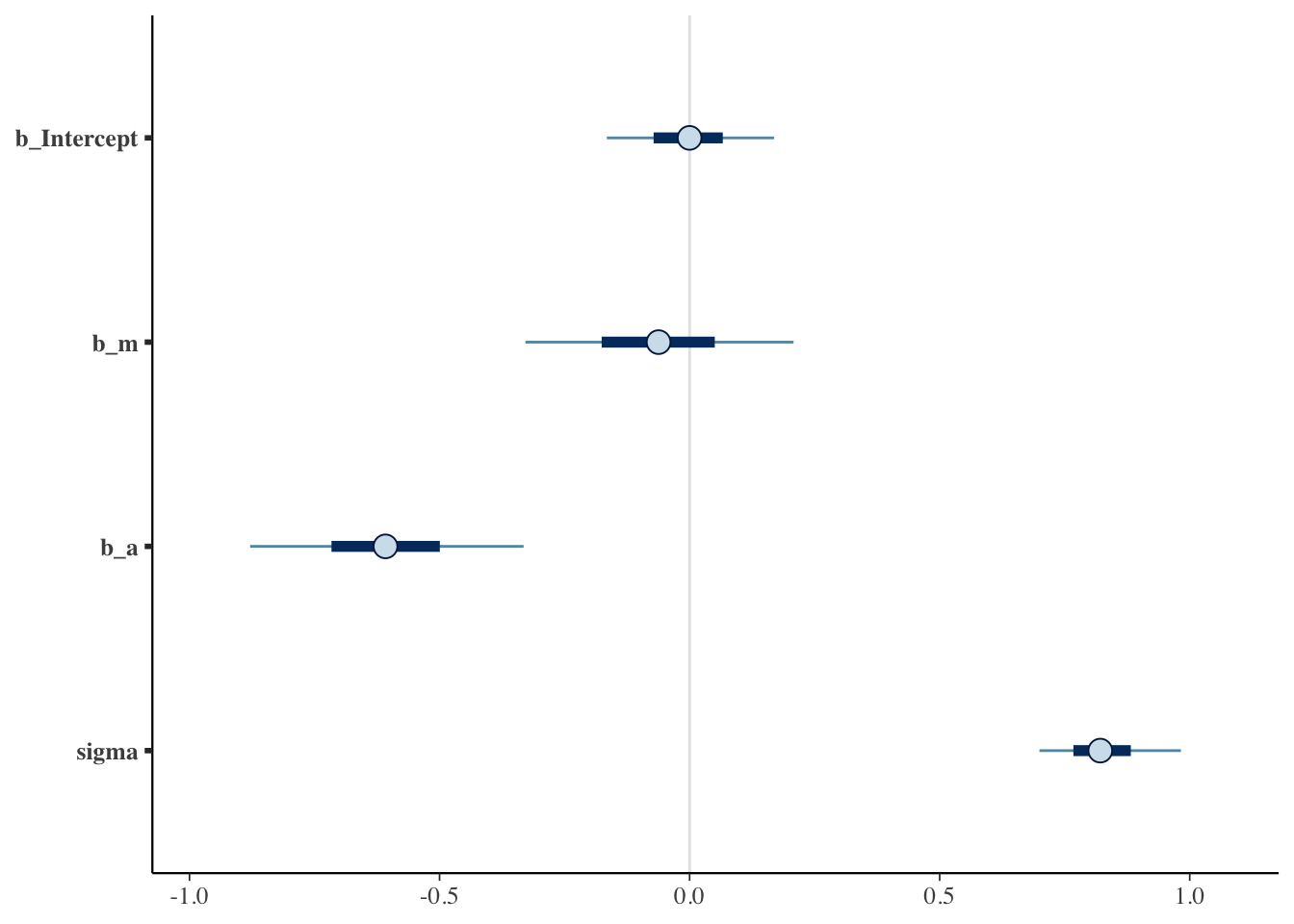
Lets look at the coefficients:
Code
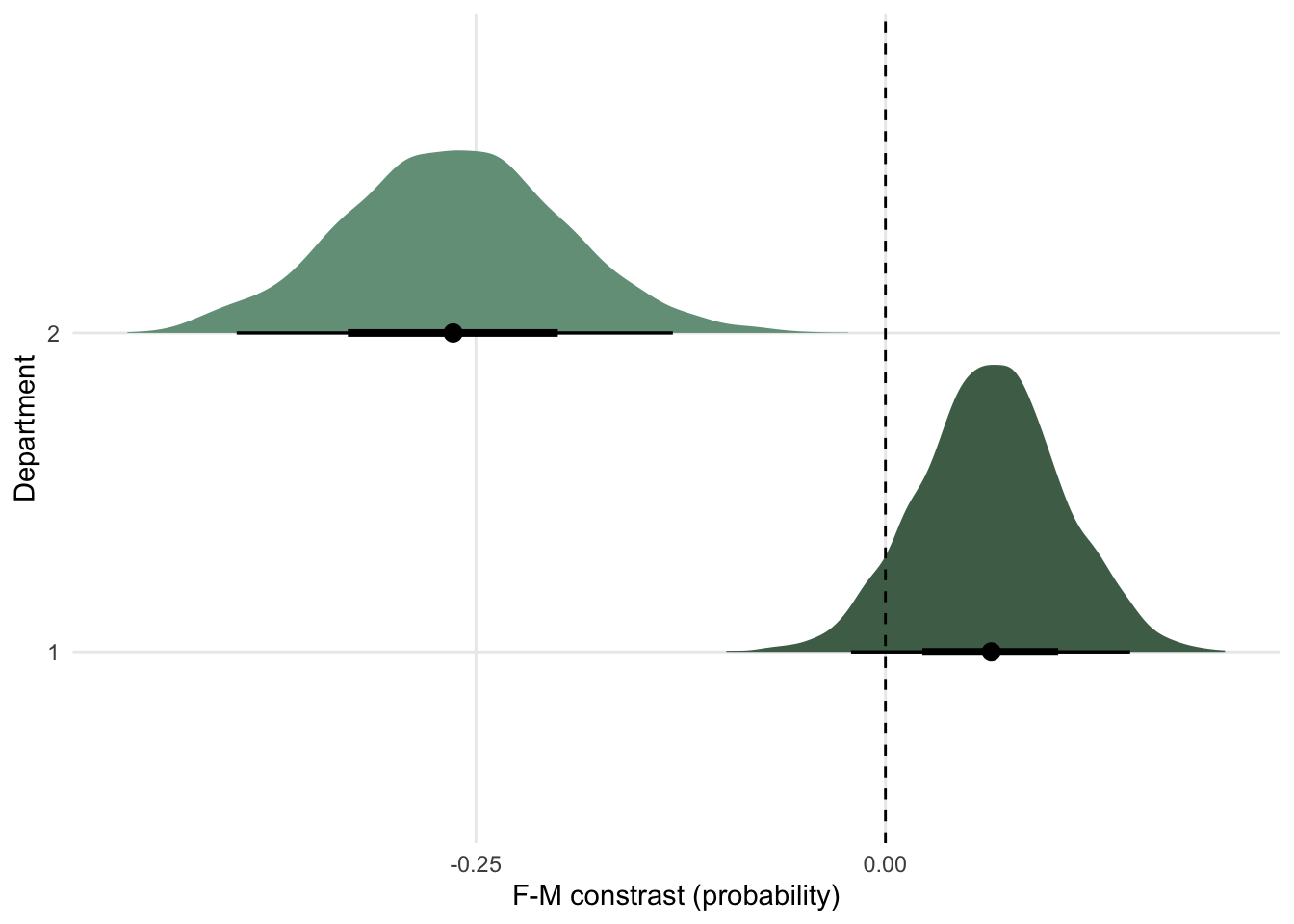
mcmc_plot(marriage_model)
There is a very small effect of marriage, but we aren’t sure if this very small effect is positive or negative. Essentially no causal effect on divorce.
Age of marriage has a large negative effect though.
\(~\)
Gold standard: simulating an intervention from the data
Code
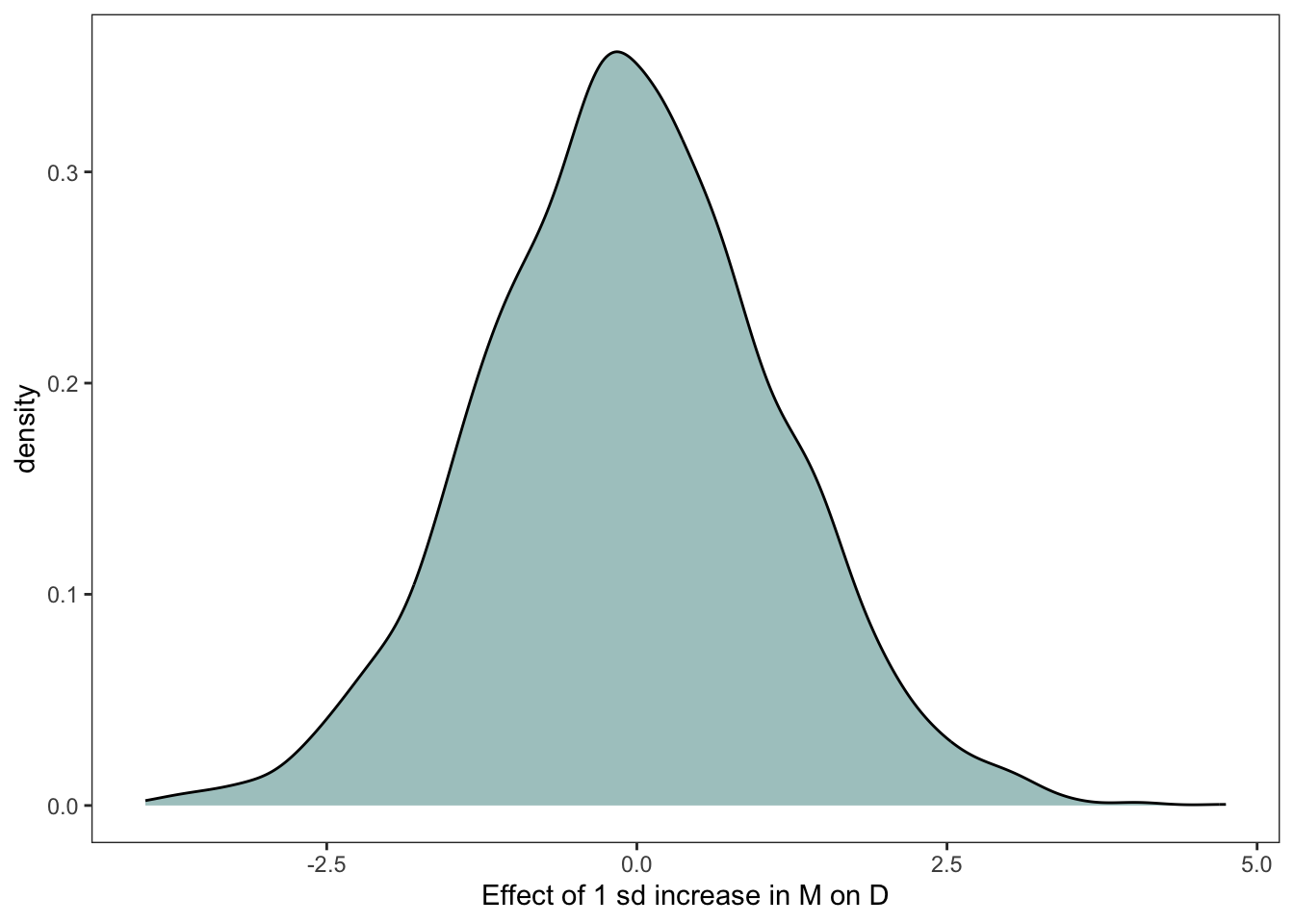
post <-as_draws_df(marriage_model)n <-4000A <-sample(Waffle_data$a, size = n, replace = T)# simulate divorce for M = 0 bind_cols(post %>%mutate(m_0 =rnorm(n, b_Intercept + b_m *0+ b_a * A, sigma)) %>%select(m_0),# now 1 sd above the meanpost %>%mutate(m_1 =rnorm(n, b_Intercept + b_m *1+ b_a * A, sigma)) %>%select(m_1)) %>%mutate(diff = m_1 - m_0) %>%ggplot(aes(x = diff)) +geom_density(fill =met.brewer("Hokusai2")[1]) +labs(x ="Effect of 1 sd increase in M on D") +#coord_cartesian(ylim = c(-2, 2)) +theme_bw() +theme(panel.grid =element_blank())
\(~\)
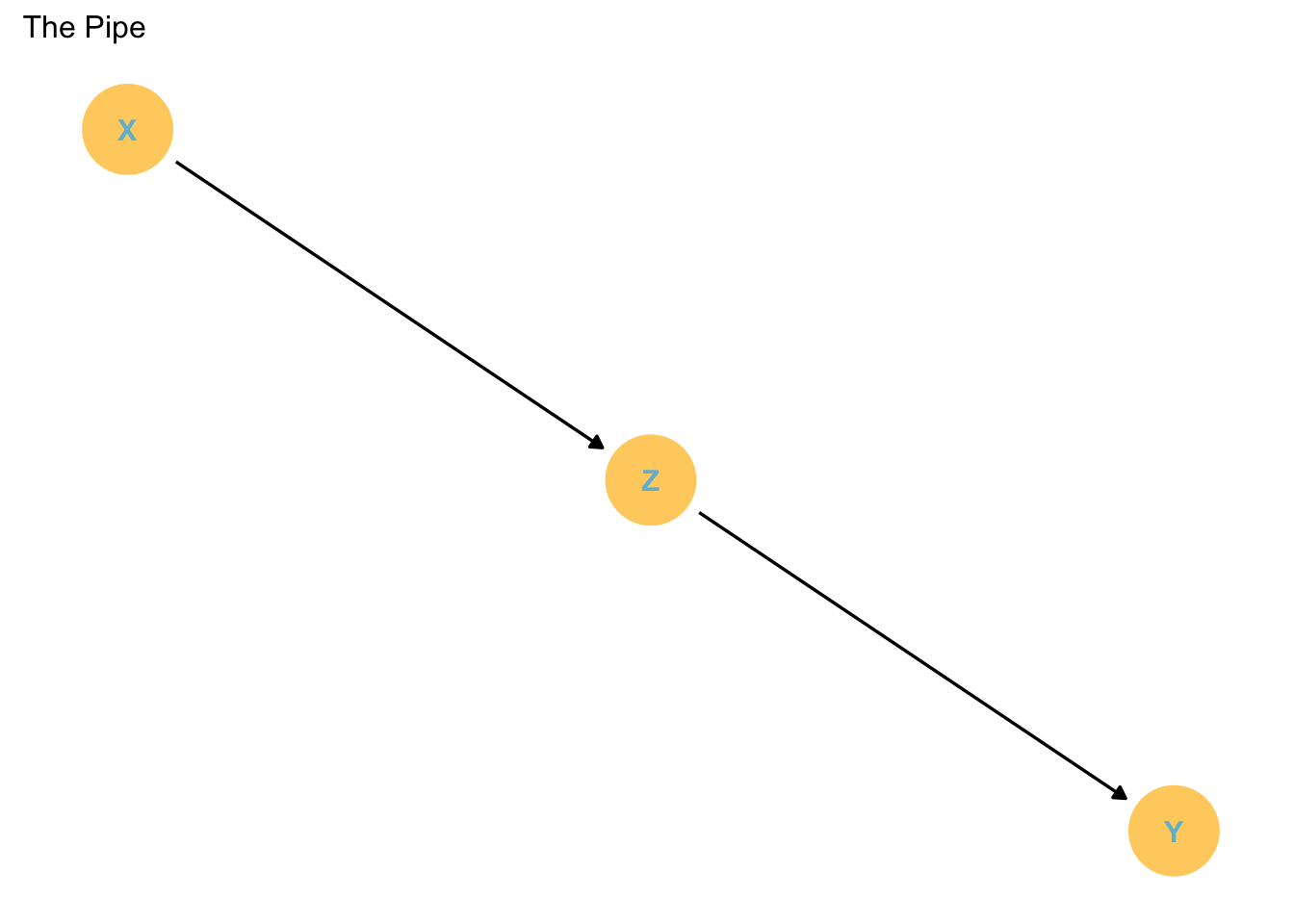
The Pipe
\(~\)
Structurally different to the fork, but statistically handled similarly.
In this case Z is a mediator variable. That is, the influence of X on Y is transmitted through Z.
Code
p2
Simulate to understand. Look carefully, we don’t code any effect of X on Y or vice-versa.
Code
n <-1000tibble(X =rbernoulli(n, 0.5)) %>%mutate(X =if_else(X =="TRUE", 1, 0),Z =rbernoulli(n, (1- X) *0.1+ X *0.9),Y =rbernoulli(n, (1- Z)*0.1+ Z *0.9),Z =if_else(X =="TRUE", 1, 0),Y =if_else(Y =="TRUE", 1, 0)) %>%count(X, Y)
# A tibble: 4 × 3
X Y n
<dbl> <dbl> <int>
1 0 0 431
2 0 1 90
3 1 0 111
4 1 1 368
Yet there appears to be an effect! This is because X effects Z which affects Y.
An example: a plant growth experiment
Create the data
Code
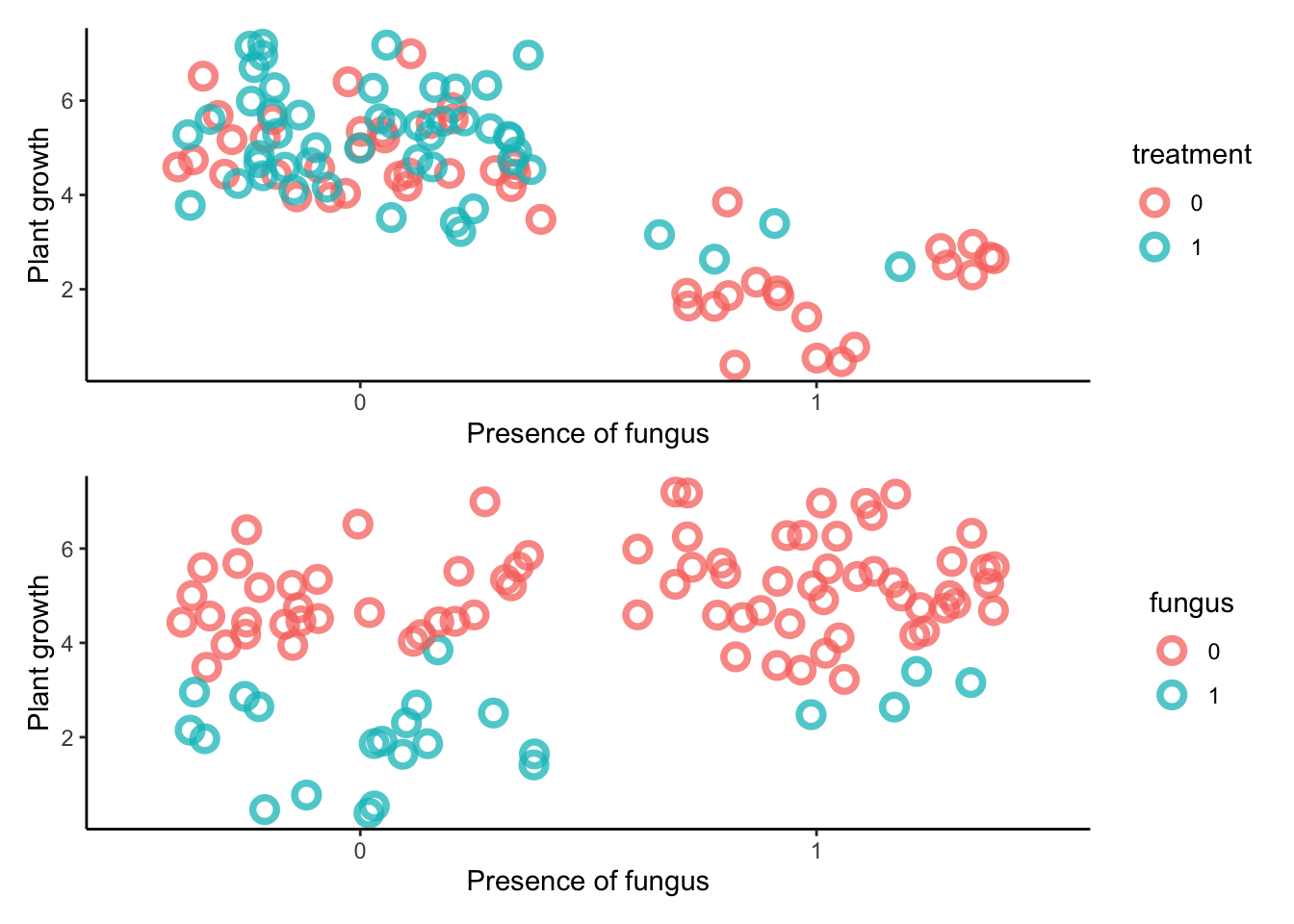
# how many plants would you like?n <-100# h0 is the starting height# treatment is an antifungal# fungus is fungus presence# h1 is end of experiment plant heightset.seed(71)plant_experiment <-tibble(h0 =rnorm(n, mean =10, sd =2), treatment =rep(0:1, each = n /2),fungus =rbinom(n, size =1, prob = .5- treatment *0.4),h1 = h0 +rnorm(n, mean =5-3* fungus, sd =1)) %>%mutate(treatment =as.factor(treatment),fungus =as.factor(fungus))
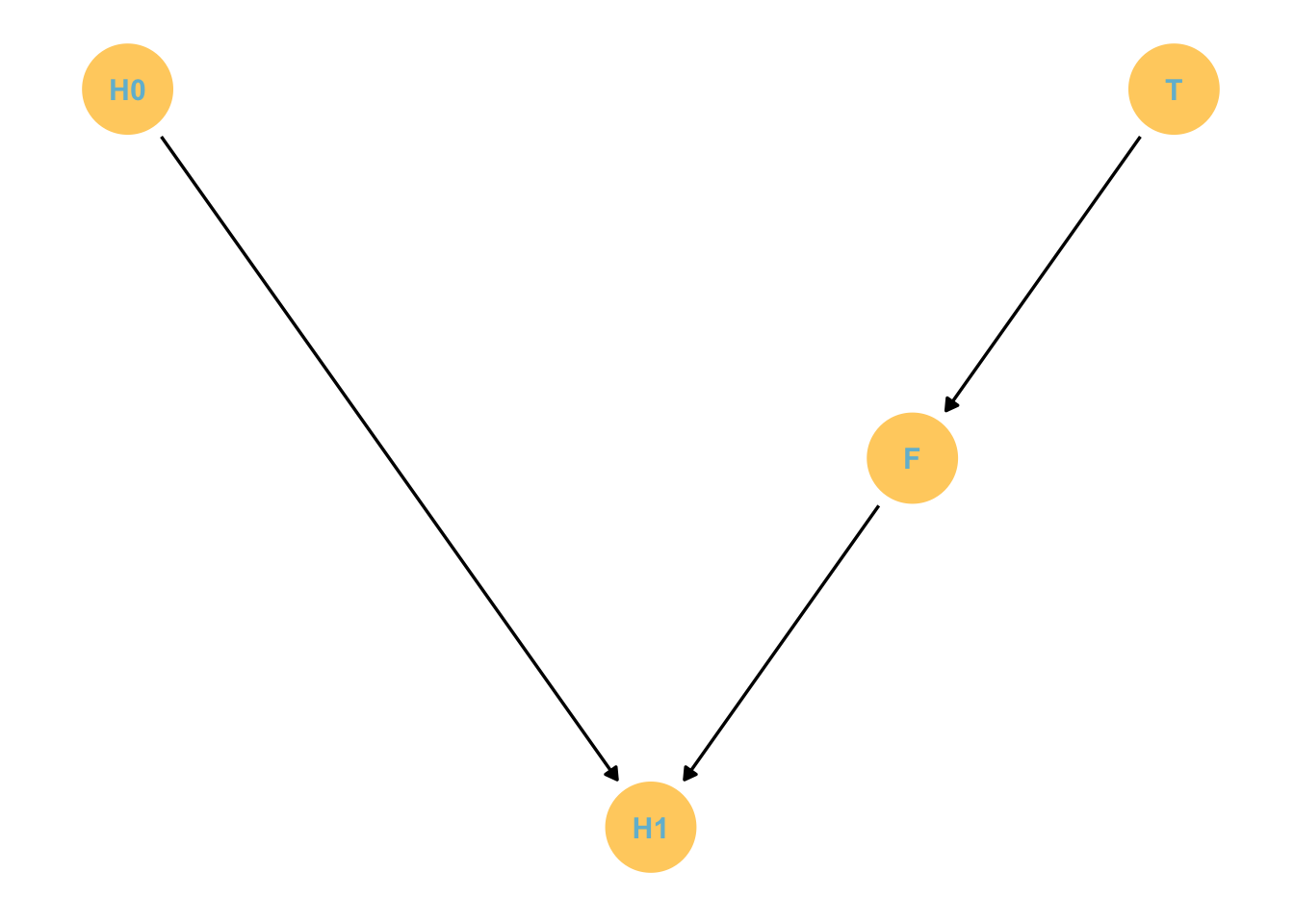
There are 100 plants troubled by fungal growth. Half the plants receive anti-fungal treatments. We measure growth and presence of fungus.
The estimand is the effect of anti-fungal treatment on growth.
The correct stats analysis here is to ignore the fungal status. This is because this will block the pipe! We remove the effect of the desired part of the experiment.
Family: gaussian
Links: mu = identity; sigma = identity
Formula: h1 ~ 0 + h0 + treatment + fungus
Data: plant_experiment (Number of observations: 100)
Draws: 4 chains, each with iter = 2000; warmup = 1000; thin = 1;
total post-warmup draws = 4000
Population-Level Effects:
Estimate Est.Error l-95% CI u-95% CI Rhat Bulk_ESS Tail_ESS
h0 1.34 0.03 1.28 1.41 1.00 1371 1559
treatment0 1.00 0.36 0.28 1.68 1.00 1468 2101
treatment1 1.46 0.37 0.71 2.16 1.00 1481 2258
fungus1 -1.73 0.29 -2.28 -1.15 1.00 2674 2344
Family Specific Parameters:
Estimate Est.Error l-95% CI u-95% CI Rhat Bulk_ESS Tail_ESS
sigma 1.33 0.11 1.13 1.57 1.00 1784 2131
Draws were sampled using sampling(NUTS). For each parameter, Bulk_ESS
and Tail_ESS are effective sample size measures, and Rhat is the potential
scale reduction factor on split chains (at convergence, Rhat = 1).
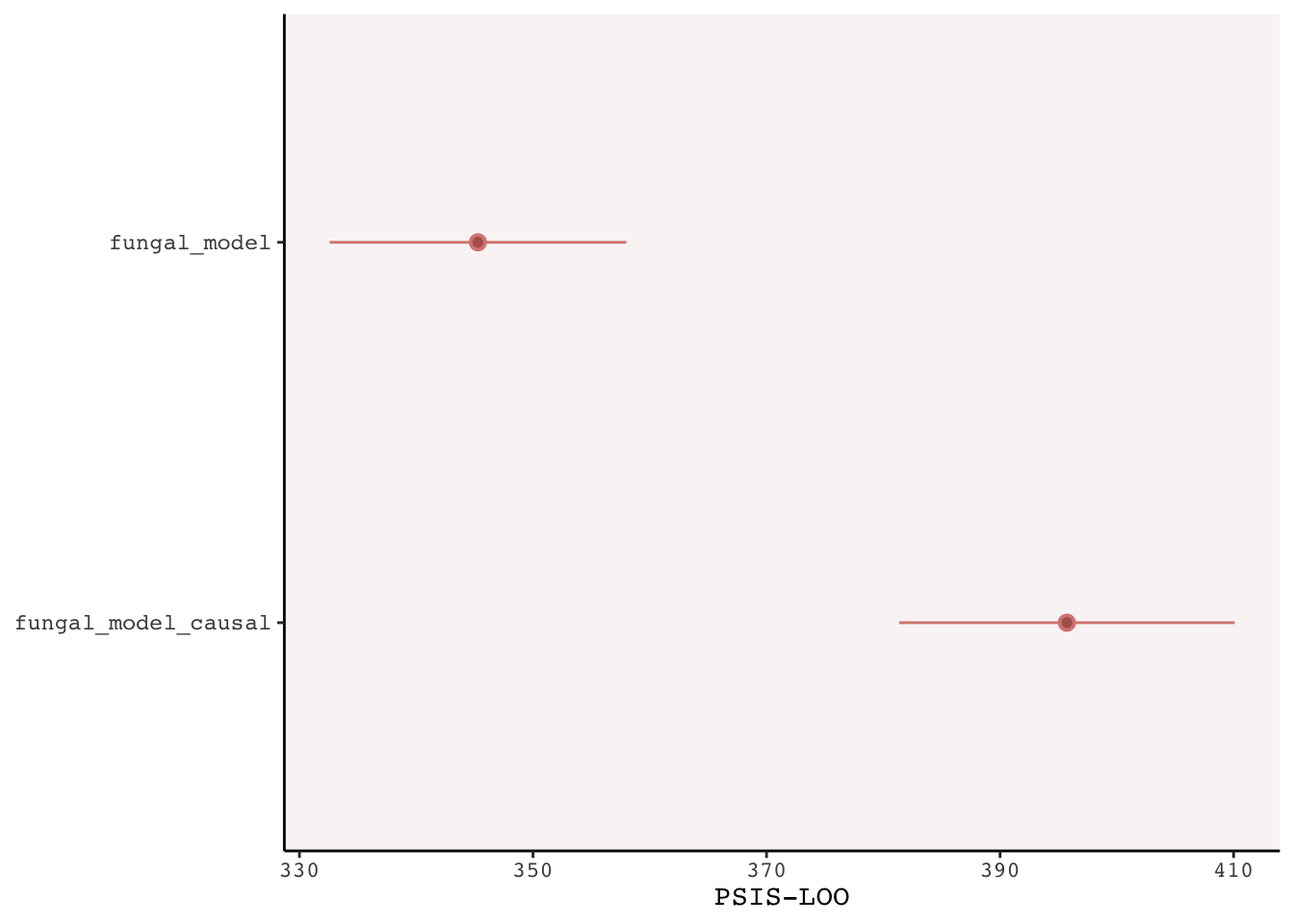
The treatment works through fungal growth, so if we account for fungal growth, of course we will find no effect. So the message is not to stratify by an effect of the treatment you’re interested in.
To prove this, lets fit the more logical model:
Code
fungal_model_causal <-brm(data = plant_experiment, family = gaussian, h1 ~0+ h0 + treatment,prior =c(prior(normal(0, 0.5), class = b),prior(exponential(1), class = sigma)),iter =2000, warmup =1000, chains =4, cores =4,seed =5,file ="fits/fungal_model_causal")fungal_model_causal
Family: gaussian
Links: mu = identity; sigma = identity
Formula: h1 ~ 0 + h0 + treatment
Data: plant_experiment (Number of observations: 100)
Draws: 4 chains, each with iter = 2000; warmup = 1000; thin = 1;
total post-warmup draws = 4000
Population-Level Effects:
Estimate Est.Error l-95% CI u-95% CI Rhat Bulk_ESS Tail_ESS
h0 1.34 0.04 1.27 1.41 1.00 1293 1603
treatment0 -0.01 0.37 -0.75 0.69 1.00 1454 1903
treatment1 1.04 0.38 0.30 1.79 1.00 1338 1618
Family Specific Parameters:
Estimate Est.Error l-95% CI u-95% CI Rhat Bulk_ESS Tail_ESS
sigma 1.72 0.12 1.49 1.98 1.00 2100 2128
Draws were sampled using sampling(NUTS). For each parameter, Bulk_ESS
and Tail_ESS are effective sample size measures, and Rhat is the potential
scale reduction factor on split chains (at convergence, Rhat = 1).
You should be very wary of including consequences of your treatment other than the desired outcome variable because of the pipe.
\(~\)
The Collider
\(~\)
Code
p3
X and Y are not associated but both influence Z.
If you stratify by Z, then X and Y become associated! How??
If we learn the value of Z we have to learn something about X and Y. Still confused?
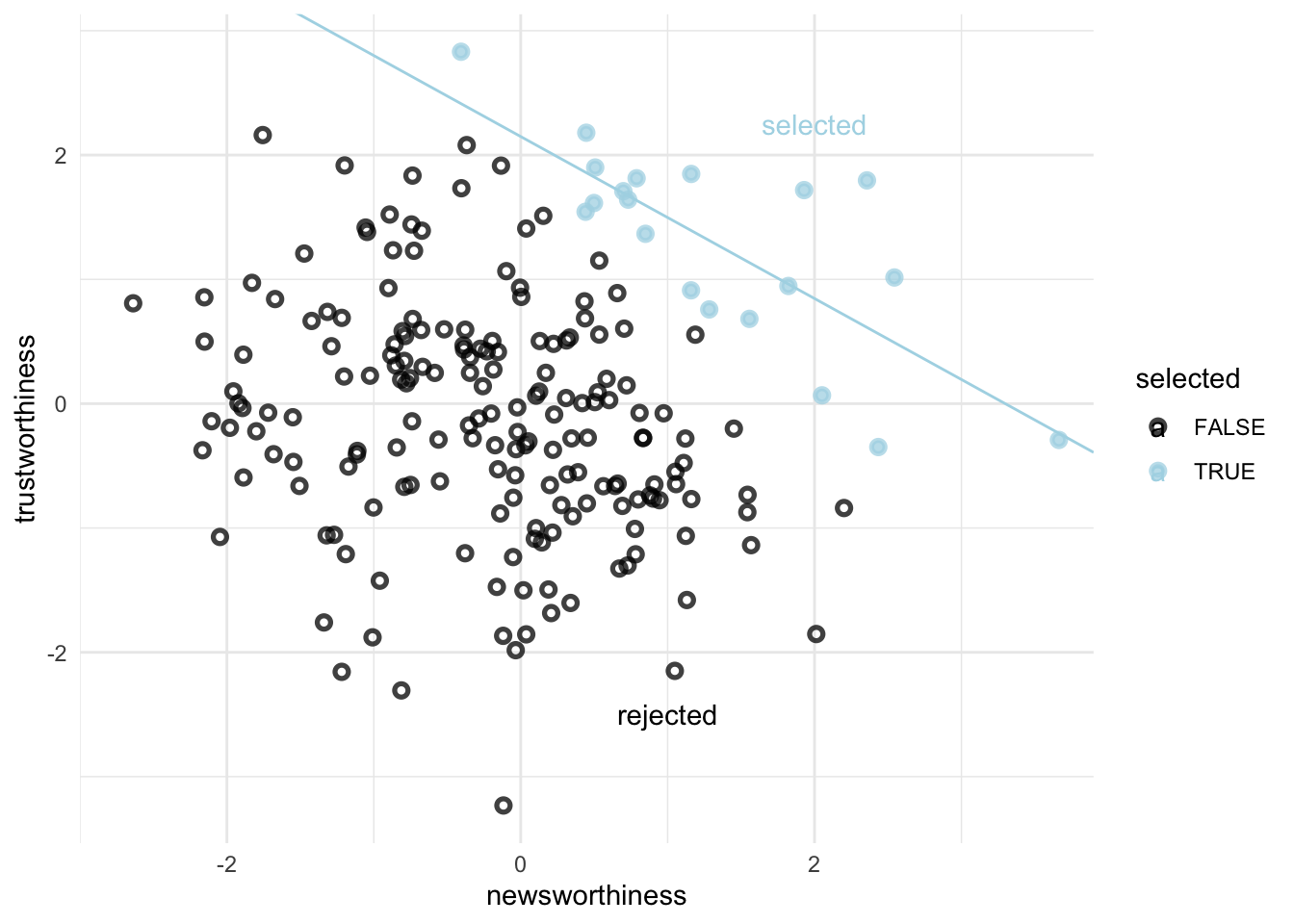
An example: Selection bias in grants
There are 200 grant applications
Rated on trustworthiness and newsworthiness
These are uncorrelated variables
Load in the data and plot the relationship
Code
set.seed(1914)n <-200# number of grant proposalsp <-0.1# proportion to selectgrants <-# uncorrelated newsworthiness and trustworthinesstibble(newsworthiness =rnorm(n, mean =0, sd =1),trustworthiness =rnorm(n, mean =0, sd =1)) %>%# total_scoremutate(total_score = newsworthiness + trustworthiness) %>%# select top 10% of combined scoresmutate(selected =ifelse(total_score >=quantile(total_score, 1- p), TRUE, FALSE))
Code
# we'll need this for the annotationtext <-tibble(newsworthiness =c(2, 1), trustworthiness =c(2.25, -2.5),selected =c(TRUE, FALSE),label =c("selected", "rejected"))grants %>%ggplot(aes(x = newsworthiness, y = trustworthiness, color = selected)) +geom_point(aes(shape = selected), alpha =3/4, stroke =1.4) +geom_text(data = text,aes(label = label)) +geom_smooth(data = . %>%filter(selected ==TRUE),method ="lm", fullrange = T,color ="lightblue", se = F, size =1/2) +scale_color_manual(values =c("black", "lightblue")) +scale_shape_manual(values =c(1, 19)) +scale_x_continuous(limits =c(-3, 3.9), expand =c(0, 0)) +coord_cartesian(ylim =range(grants$trustworthiness)) +theme(legend.position ="none") +theme_minimal()
After selection there is a strong negative correlation.
Ok, so due to selection the only grants that make it are either high in newsworthiness or trustworthiness (makes sense). From a random distribution few grants will be high in both (this is much rarer than being high in one of the categories), The result is a subset of grants that are good at one thing but mainly bad at the other, which creates the appearance of a negative correlation across the entire sample. The key is to not subset the data (don’t stratify)
Example 2: selection bias in restaurants
Restaurants are successful because they have good food or because they’re in a good location.
Only the bad restaurants in really good locations can survive.
Creates a negative relationship between food quality and location.
Example 3: selection bias in actors
Actors can be successful because they are skilled or because they are good looking.
Really bad actors can survive if they are attractive - they end up being the only less skilled actors that can survive.
Selection creates a negative relationship between looks and skill.
\(~\)
Stats example: endogenous colliders
If you condition on a collider, you create phantom non-causal associations.
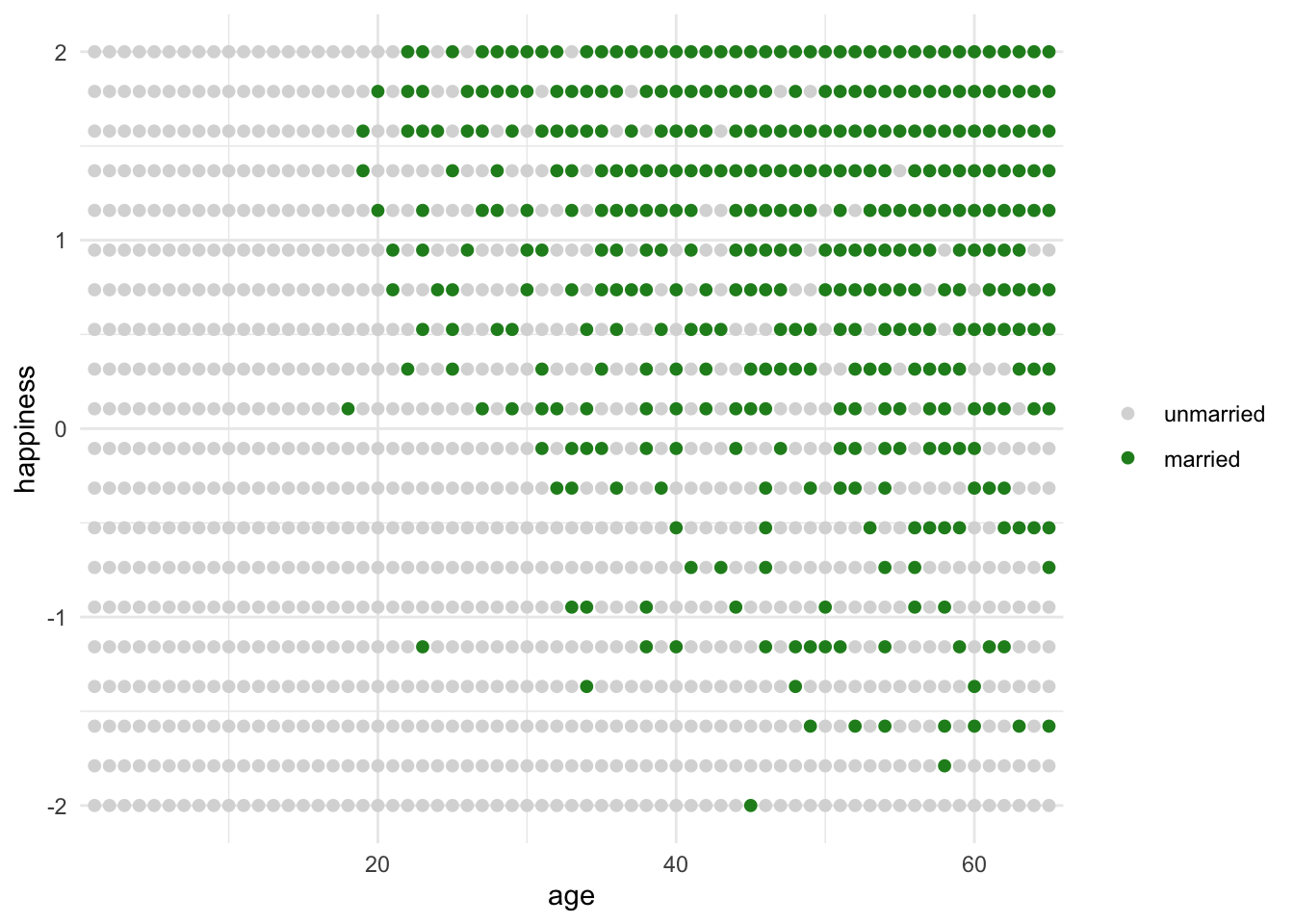
Suppose age has no effect on happiness but both affect marital status.
So the collider scenario here is that of the married people, only a very advanced age can overcome unhapiness or a very happy dispoistion can overcome a very young age = a neg relationship
If the visualisation isn’t enough to convince you, lets fit the models.
First with the collider
Code
marriage_sim_2 <- marriage_sim %>%mutate(mid =factor(married +1, labels =c("single", "married"))) %>%# only inlcude those 18 and abovefilter(age >17) %>%# create a, a standarised variable where 18 = 0 and 65 = 1mutate(a = (age -18) / (65-18))marriage_happiness_model <-brm(data = marriage_sim_2, family = gaussian, happiness ~0+ mid + a,prior =c(prior(normal(0, 1), class = b, coef = midmarried),prior(normal(0, 1), class = b, coef = midsingle),prior(normal(0, 2), class = b, coef = a),prior(exponential(1), class = sigma)),iter =2000, warmup =1000, chains =4, cores =4,seed =6,file ="fits/marriage_happiness_model")marriage_happiness_model
Family: gaussian
Links: mu = identity; sigma = identity
Formula: happiness ~ 0 + mid + a
Data: marriage_sim_2 (Number of observations: 960)
Draws: 4 chains, each with iter = 2000; warmup = 1000; thin = 1;
total post-warmup draws = 4000
Population-Level Effects:
Estimate Est.Error l-95% CI u-95% CI Rhat Bulk_ESS Tail_ESS
midsingle -0.24 0.06 -0.36 -0.11 1.00 1732 2058
midmarried 1.26 0.08 1.09 1.42 1.00 1758 1943
a -0.75 0.11 -0.96 -0.53 1.00 1508 2084
Family Specific Parameters:
Estimate Est.Error l-95% CI u-95% CI Rhat Bulk_ESS Tail_ESS
sigma 0.99 0.02 0.95 1.04 1.00 2479 2033
Draws were sampled using sampling(NUTS). For each parameter, Bulk_ESS
and Tail_ESS are effective sample size measures, and Rhat is the potential
scale reduction factor on split chains (at convergence, Rhat = 1).
As we expected, age appears to be negatively associated with happiness.
Now remove the collider
Code
marriage_happiness_model_causal <-brm(data = marriage_sim_2, family = gaussian, happiness ~0+ a,prior =c(prior(normal(0, 2), class = b, coef = a),prior(exponential(1), class = sigma)),iter =2000, warmup =1000, chains =4, cores =4,seed =6,file ="fits/marriage_happiness_model_causal")marriage_happiness_model_causal
Family: gaussian
Links: mu = identity; sigma = identity
Formula: happiness ~ 0 + a
Data: marriage_sim_2 (Number of observations: 960)
Draws: 4 chains, each with iter = 2000; warmup = 1000; thin = 1;
total post-warmup draws = 4000
Population-Level Effects:
Estimate Est.Error l-95% CI u-95% CI Rhat Bulk_ESS Tail_ESS
a -0.00 0.07 -0.13 0.14 1.00 3281 2571
Family Specific Parameters:
Estimate Est.Error l-95% CI u-95% CI Rhat Bulk_ESS Tail_ESS
sigma 1.21 0.03 1.16 1.27 1.00 3192 2242
Draws were sampled using sampling(NUTS). For each parameter, Bulk_ESS
and Tail_ESS are effective sample size measures, and Rhat is the potential
scale reduction factor on split chains (at convergence, Rhat = 1).
Age has literally no effect on happiness now, as we specified in the simulation.
\(~\)
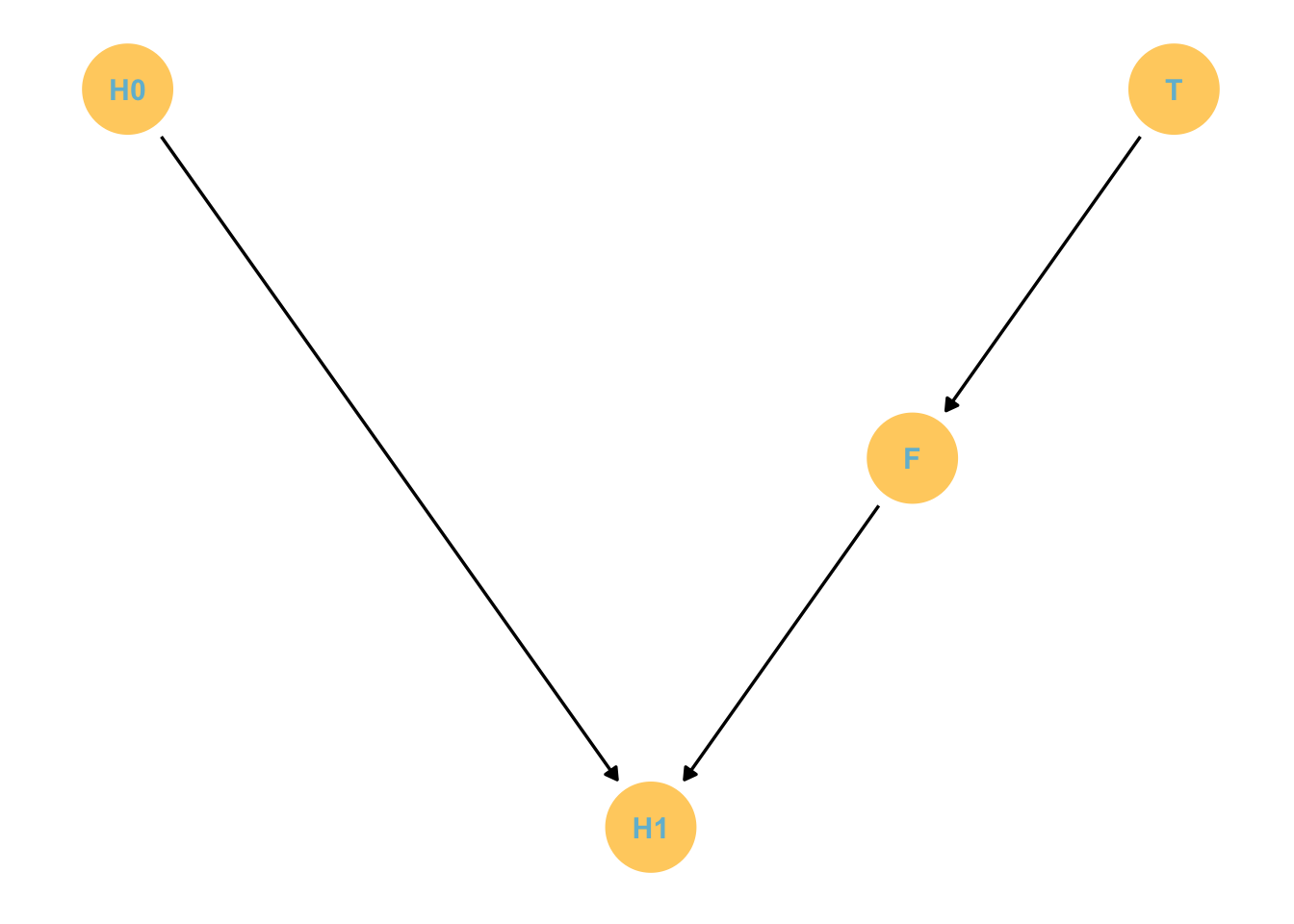
The Descendant
\(~\)
Code
d4 <-dagify(Z ~ X + Y, D ~ Z,coords =tibble(name =c("X", "Y", "Z", "D"),x =c(1, 3, 2, 2),y =c(1, 1, 2, 1.05)) )p4 <-gg_simple_dag(d4) +labs(subtitle ="The Descendant")p4
The variable A is the descendant of the Z variable.
The consequence is if we condition (stratify) by A it will have the same effect as conditioning by Z. While A does not affect X or Y, it is related to Z so it has a weaker (depending on the A -> association strength) but tangible effect on X/Y.
Think about how common descendants are. Any proxy is a descendant!
E.g. all fitness measures are proxies and therefore descendants of true fitness.
\(~\)
Lecture 6: Do calculus and good and bad controls
\(~\)
Unobserved confounds
\(~\)
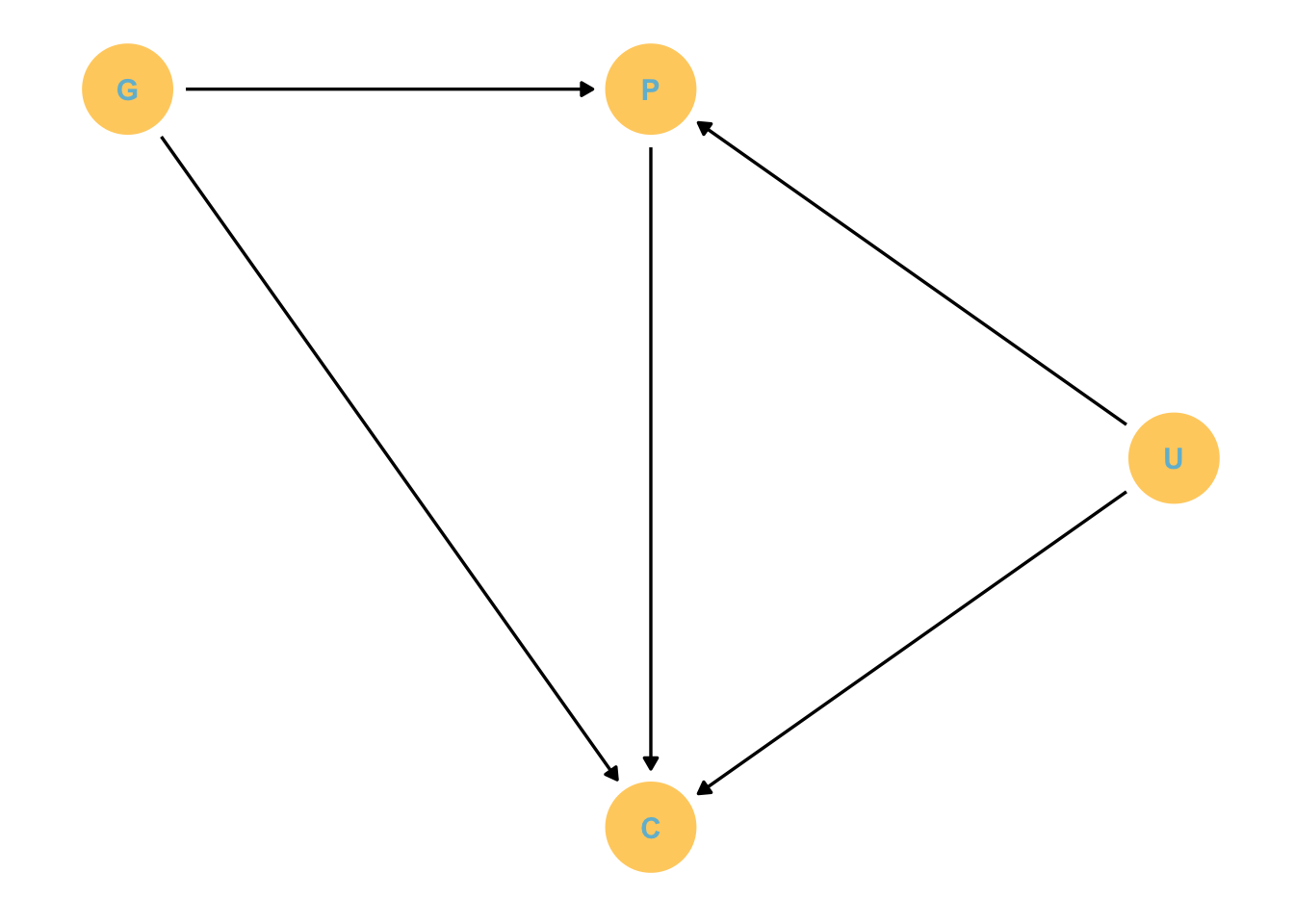
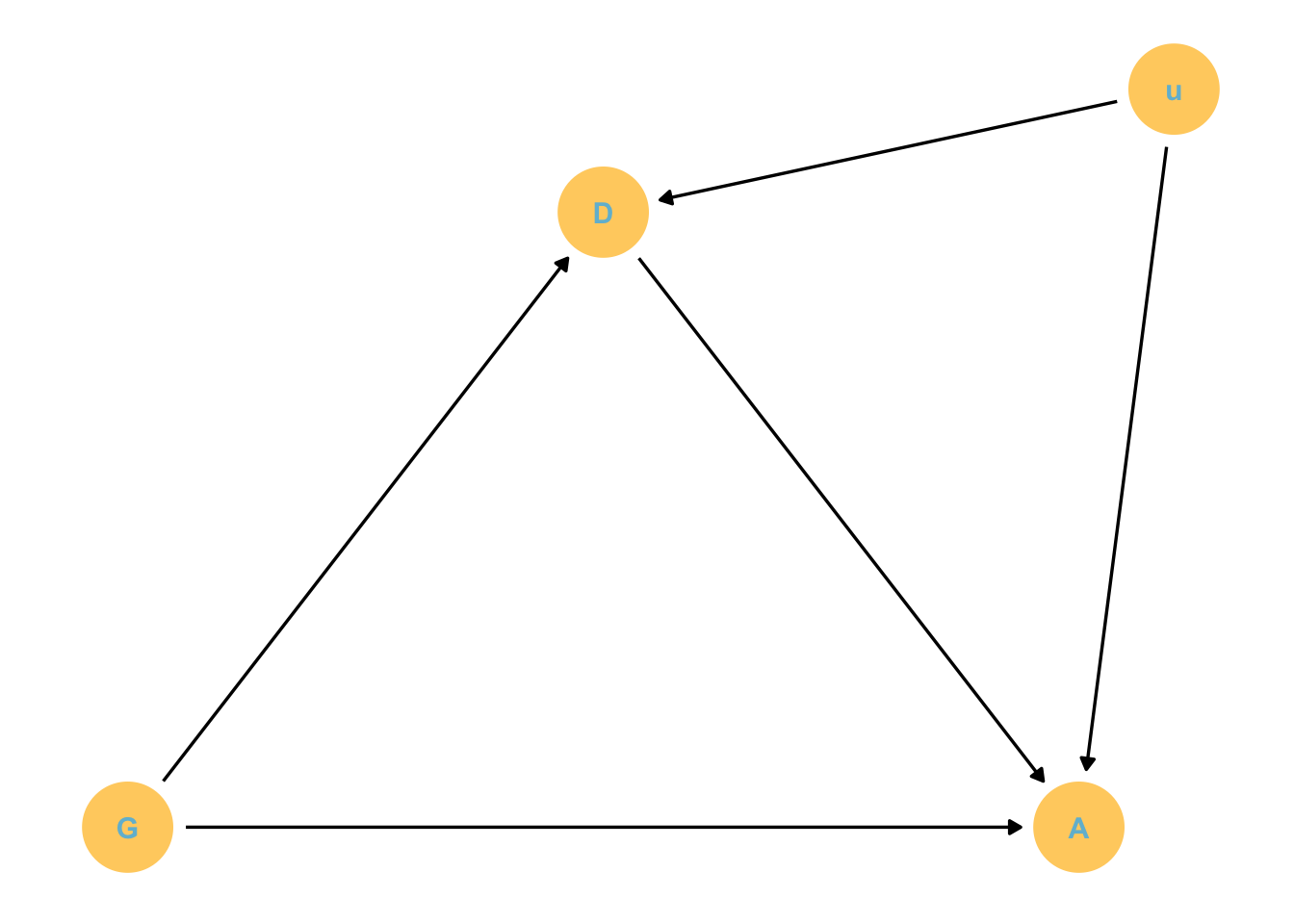
Suppose we are interested in the direct effect of grandparents education on their grandchildrens education.
They will have direct and indirect effects on these kids
Estimand: Direct effect of grandparents G on children C.
But parent education is obviously an issue here.
Code
dag_coords <-tibble(name =c("G", "P", "C", "U"),x =c(1, 2, 2, 3),y =c(2, 2, 1, 1.5))dagify(P ~ G + U, C ~ P + G + U,coords = dag_coords) %>%gg_simple_dag()
The parental effect is likely confounded with living conditions. However, this might not be the case for grandparents who live in a different area.
Remember what our goal is: estimate the direct causal effect of grandparents.
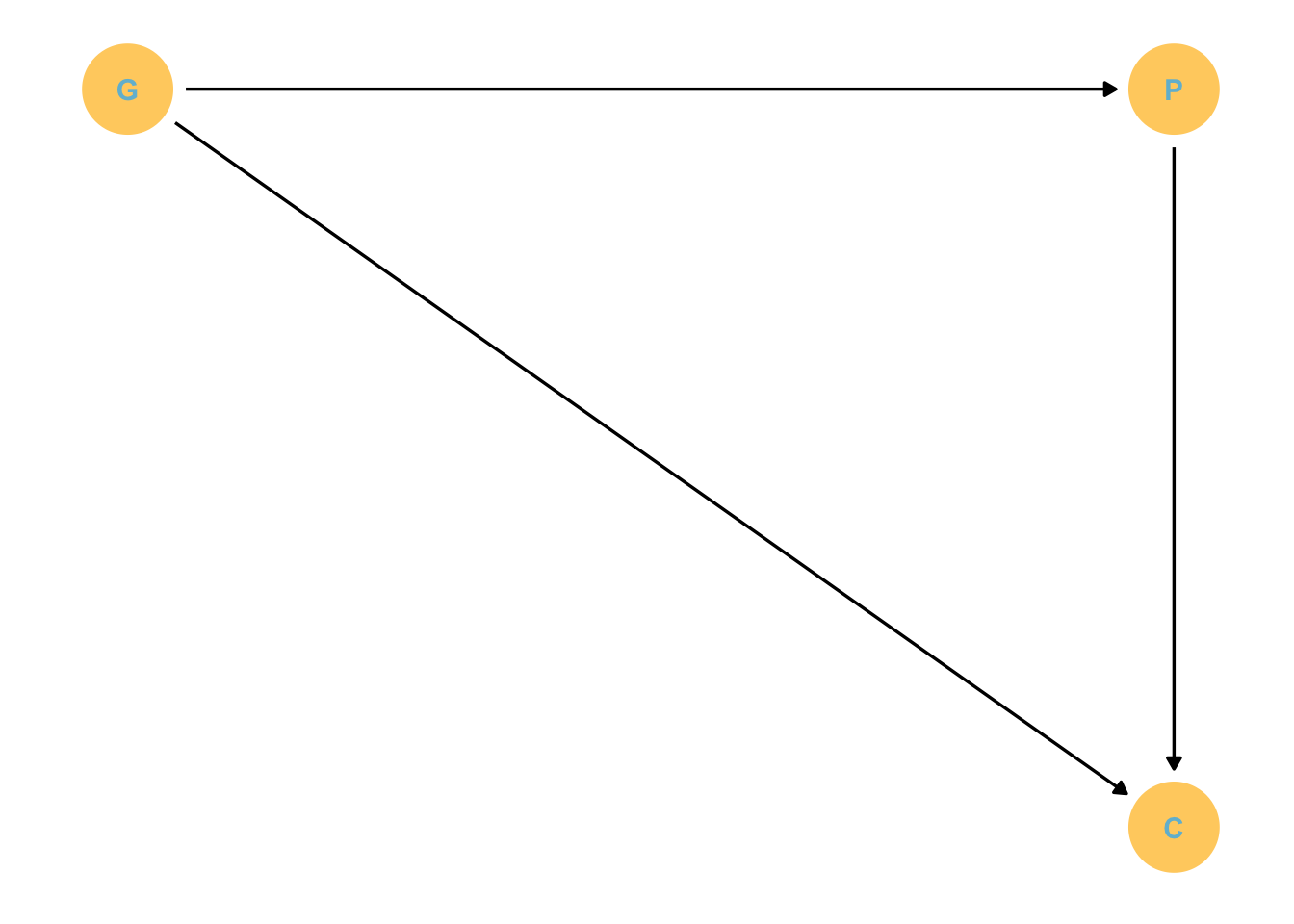
If the situation was this:
Code
dagify(P ~ G, C ~ P + G,coords = dag_coords) %>%gg_simple_dag()
Then we would just have to stratify by P to block the pipe and find the direct effect of G.
Unfortunately, U exists, which makes P a collider! So this makes P potentially associated with C because U affects them both.
We are left with only bad choices. Sometimes this is the way it is.
Why does collider bias happen: continuing with the parental example.
Let’s simulate the data, with G having no causal effect on C.
Code
# how many grandparent-parent-child triads would you like?n <-200b_gp <-1# direct effect of G on Pb_gc <-0# direct effect of G on Cb_pc <-1# direct effect of P on Cb_u <-2# direct effect of U on P and C# simulate triadsset.seed(1)d <-tibble(u =2*rbinom(n, size =1, prob = .5) -1,g =rnorm(n, mean =0, sd =1)) %>%mutate(p =rnorm(n, mean = b_gp * g + b_u * u, sd =1)) %>%mutate(c =rnorm(n, mean = b_pc * p + b_gc * g + b_u * u, sd =1))
Now we’ll fit the model without U - which closes the pipe but opens the collider.
Code
parent_collider_model <-brm(data = d, family = gaussian, c ~0+ Intercept + p + g,prior =c(prior(normal(0, 1), class = b),prior(exponential(1), class = sigma)),iter =2000, warmup =1000, chains =4, cores =4,seed =6,file ="fits/parent_collider_model")parent_collider_model
Family: gaussian
Links: mu = identity; sigma = identity
Formula: c ~ 0 + Intercept + p + g
Data: d (Number of observations: 200)
Draws: 4 chains, each with iter = 2000; warmup = 1000; thin = 1;
total post-warmup draws = 4000
Population-Level Effects:
Estimate Est.Error l-95% CI u-95% CI Rhat Bulk_ESS Tail_ESS
Intercept -0.12 0.10 -0.31 0.07 1.00 4158 2829
p 1.79 0.05 1.70 1.87 1.00 3550 3015
g -0.84 0.11 -1.04 -0.63 1.00 3927 2951
Family Specific Parameters:
Estimate Est.Error l-95% CI u-95% CI Rhat Bulk_ESS Tail_ESS
sigma 1.43 0.07 1.30 1.57 1.00 4142 2926
Draws were sampled using sampling(NUTS). For each parameter, Bulk_ESS
and Tail_ESS are effective sample size measures, and Rhat is the potential
scale reduction factor on split chains (at convergence, Rhat = 1).
Lets plot what’s happening
Code
d %>%mutate(centile =ifelse(p >=quantile(p, prob = .45) & p <=quantile(p, prob = .60), "a", "b"),u =factor(u)) %>%ggplot(aes(x = g, y = c)) +geom_point(aes(shape = centile, color = u),size =2.5, stroke =1/4) +stat_smooth(data = . %>%filter(centile =="a"),method ="lm", se = F, size =1/2, color ="black", fullrange = T) +scale_shape_manual(values =c(19, 1)) +scale_color_manual(values =c("black", "lightblue")) +theme(legend.position ="none")
Stratifying by P as the regression line shows creates a negative relationship where nothing causal is happening!
Now we’ll fit the model with U
Code
grandparent_total <-update(parent_collider_model,newdata = d,formula = c ~0+ Intercept + p + g + u,seed =6,file ="fits/grandparent_total")grandparent_total
Family: gaussian
Links: mu = identity; sigma = identity
Formula: c ~ Intercept + p + g + u - 1
Data: d (Number of observations: 200)
Draws: 4 chains, each with iter = 2000; warmup = 1000; thin = 1;
total post-warmup draws = 4000
Population-Level Effects:
Estimate Est.Error l-95% CI u-95% CI Rhat Bulk_ESS Tail_ESS
Intercept -0.12 0.07 -0.26 0.03 1.00 3264 2707
p 1.01 0.07 0.88 1.15 1.00 1603 2175
g -0.04 0.10 -0.24 0.15 1.00 2016 2141
u 1.99 0.15 1.70 2.28 1.00 1621 2264
Family Specific Parameters:
Estimate Est.Error l-95% CI u-95% CI Rhat Bulk_ESS Tail_ESS
sigma 1.04 0.05 0.94 1.14 1.00 2871 2527
Draws were sampled using sampling(NUTS). For each parameter, Bulk_ESS
and Tail_ESS are effective sample size measures, and Rhat is the potential
scale reduction factor on split chains (at convergence, Rhat = 1).
Note the change in the effect of G on C. Remember that we coded it to have 0 effect in our simulation!
In short: there are two ways for parents to attain their education: from G or from U. This is the classic setup for a collider.
\(~\)
DAG thinking
\(~\)
In an experiment we aim to cut all the causes of the treatment through randomisation.
But if we don’t randomise, can we overcome this with statistics? Sometimes.
First some notation:
\(P(Y|do(X)) = P(Y|?)\)
Where P(Y) is the distribution of Y
So the above means the distribution of Y conditional upon do(X) equals the distribution of Y conditional on some set of information.
do(X) means to intervene on X - to set it to some particular value.
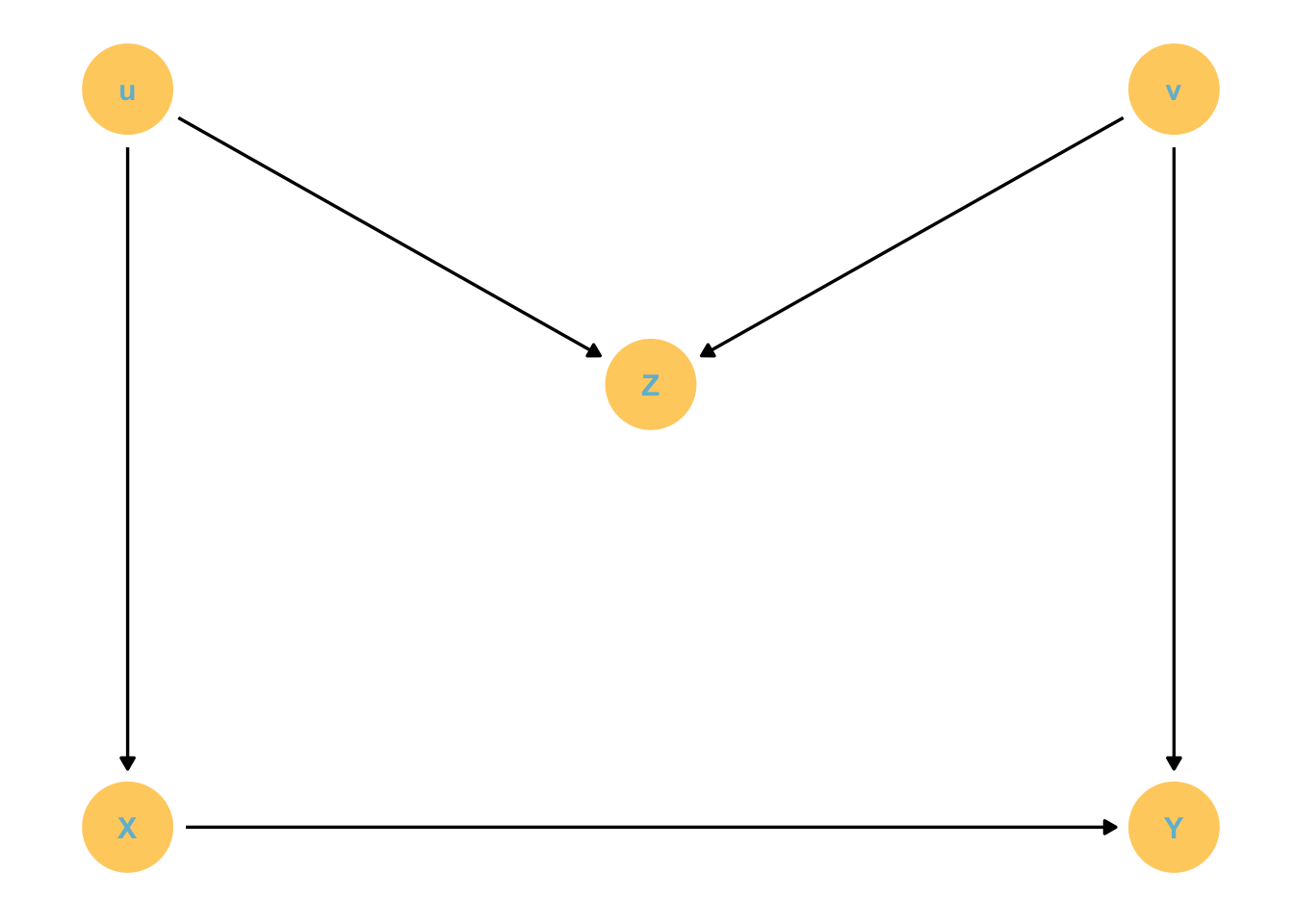
We can do this with a DAG!
Code
dag_coords <-tibble(name =c("U", "X", "Y"),x =c(2, 1, 3),y =c(2, 1, 1))dagify(X ~ U, Y ~ X + U,coords = dag_coords) %>%gg_simple_dag()
If we don’t know U we can find the distribution of Y, stratified by X and U, averaged over the distribution of U. But how?
Remember that the causal effect of X on Y is not the coefficient relating X to Y. It is actually the distribution of Y when we change X, averaged over the distributions of the control variables (U in this case). This is a marginal effect.
Do calculus
The back-door criterion
A way to analyse graphs to find sets of variables to stratify by
It can also help us design research.
Formally it is a rule to find a set of variables to stratify (condition) by to yield \(P(Y|do(X))\)
The steps:
Identify all paths connecting the treatment (X) to the outcome (Y).
Once you have the paths, focus on the paths that enter X. These are back-door paths (non-causal).
Find adjustment set (set of variables to stratify by) that closes/blocks all back-door paths.
Controls
Control variable: a variable we introduce to an analysis so that a causal estimate of something else is possible.
Things you should not do:
Add everything you’ve measured
Including all variables that aren’t collinear: sometimes both variables play a role in causality
Anything measured before treatment is fair game to add - not true
Taking some examples from Cinelli, Forney and Pearl 2021: A crash course in good and bad controls
Example 1
Code
dag_coords <-tibble(name =c("u", "X", "Z", "v", "Y"),x =c(1, 1, 2, 3, 3),y =c(2, 1, 1.6, 2, 1))dagify(X ~ u, Z ~ u + v, Y ~ v + X,coords = dag_coords) %>%gg_simple_dag()
Where u and v are unobserved variables.
Let’s say that Z denotes friendship status and X and Y the health of two people. u and v are the hobbies of person X and person Y.
If we stratify by Z, that is, include it as a control, then we open a back-door path (it is a collider). This is bad for estimating the causal effect of X on Y.
Note that Z could be a pre-treatment variable (I haven’t been taught this but apparently it’s a thing).
Do calculus time - list the paths:
X -> Y: front door
X <- u -> Z <- v -> Y: backdoor. Closed unless you include Z. A bad control.
Example 2
Code
dag_coords <-tibble(name =c("X", "Z", "u", "Y"),x =c(1, 2, 2.5, 3),y =c(1, 1, 1.2, 1))dagify(Z ~ u + X, Y ~ Z + u,coords = dag_coords) %>%gg_simple_dag()
Analyse the paths:
X -> Z -> Y: front
X -> Z <- u -> Y: still front
Z is a mediator here, so if the goal to to estimate the effect of X on Y, we should not include Z in the model anyway. There is no back-door path we have to block.
However, it’s worse than that, because u - an unexplained variable - creates a fork. Including Z makes the estimate even worse than it would usually. This should always be noted where things might have a common cause e.g. happiness and lifespan or body size and fecundity.
Add sim and plot
Note that Z would turn out to be significant. This is spurious! Don’t do backwards step selection. You need a causal model.
\(~\)
Do not touch the collider
\(~\)
DAG example
Case control bias
Code
dag_coords <-tibble(name =c("X", "Z", "Y"),x =c(1, 2, 2),y =c(1, 2, 1))dagify(Z ~ Y, Y ~ X,coords = dag_coords) %>%gg_simple_dag()
Ok, so this is a simple pipe. But if we want to know the causal effect of X on Y then we run into an issue if we control for Z.
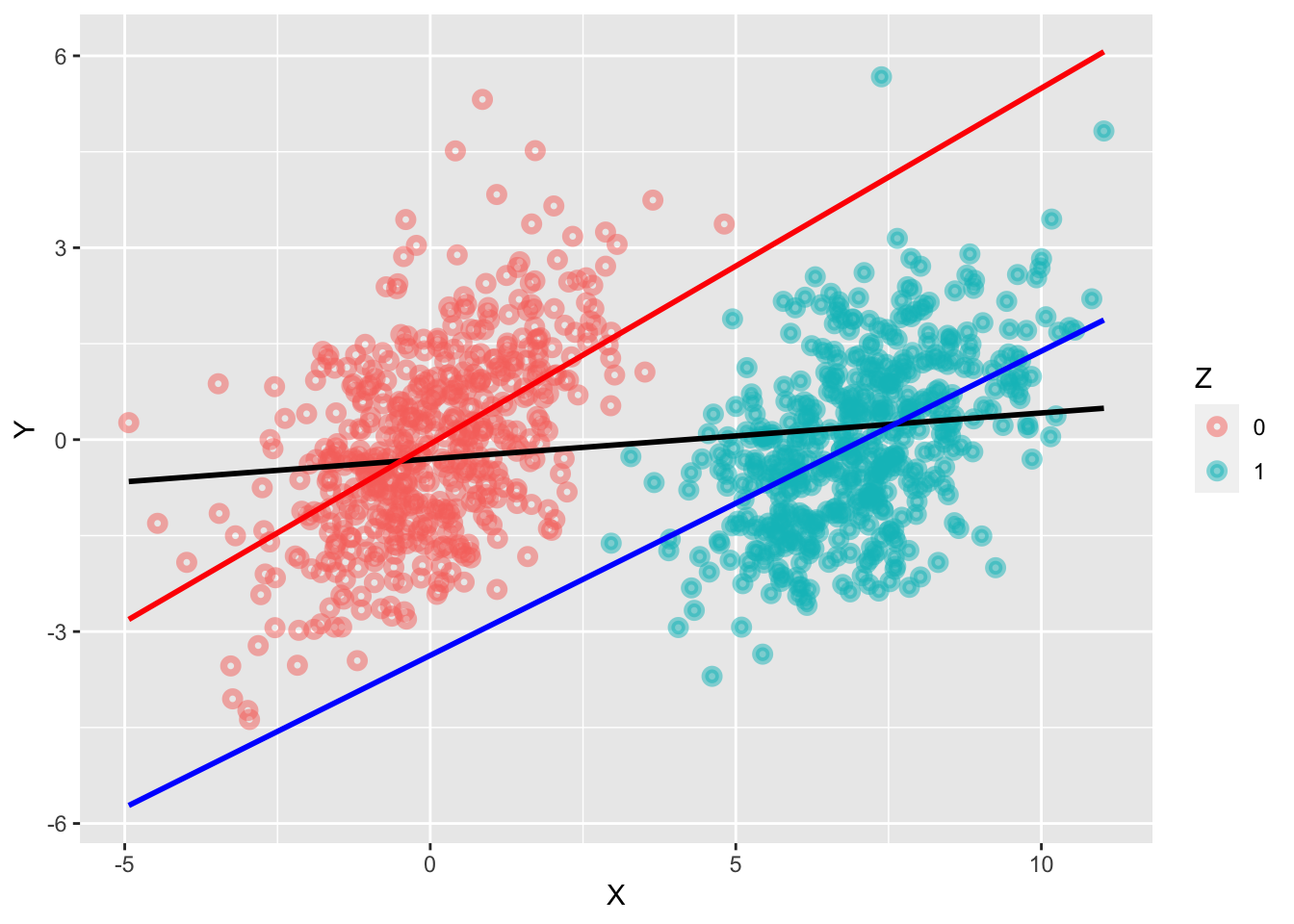
Imagine that X = education, Y = occupation and Z = Income. If we control for income, then this removes variation in Y, leaving less for education to explain - the effect size is underestimated. Now Z is not a cause of Y, but the model has no way of knowing that so it just measures the covariance. We are required to make this distinction.
A common bad control. When you apply selection on the outcome - this ruins scientific influence.
E.g. if you want to know the effect of education on occupation then you should not stratify by income because it narrows the range of cases you compare at each level.
While Z affects X it is not a back-door path because it does not connect to Y (except via the front door). However, it is still not good to condition on Z.
The parasite does not change the mean estimate, but it creates an estimate with higher variance. We lose precision.
Another sim
\(~\)
Bias amplification
A bad version of the precision parasite.
Code
dag_coords <-tibble(name =c("X", "Z", "Y", "u"),x =c(1, 1, 2, 1.5),y =c(1, 2, 1, 1.75))dagify(X ~ Z + u, Y ~ X + u,coords = dag_coords) %>%gg_simple_dag()
Remember u is an unmeasured variable. We can’t get an unbiased causal inference in this scenario.
If we add Z, the world implodes. We get a really biased estimate, with an inflated effect size.
WHY?
Covariation between X and Y requires variation in their causes. Variation is removed by conditioning - we are accounting for this association then finding the variance explained by the remaining variables. So stratifying on X removes variation in X, which gives more weight to be explained by the confound U = stronger false relationship!
Hard to get my head around but wow.
Lets plot to help understanding.
We’ll sim a situation like the above. Z is a binomial variable that causally affects X. X has no effect on Y. However, u affects both X and Y.
If Z doesn’t vary then X can’t vary. Stratifying by this removes variance in X and in that way Y. This means a larger proportion of the variation in X comes from the confounding fork u.
\(~\)
The Table 2 fallacy
\(~\)
In many fields the typical Table 2 in a manuscript will contain model coefficients.
The thing is, not all the coefficients are causal effects and there is no information that tells you whether they are.
Remember you have designed your model to identify the causal effect of X on Y. It is dangerous to interpret the effects of other variables that you have not designed the model for. The other variables are there to block back-door paths.
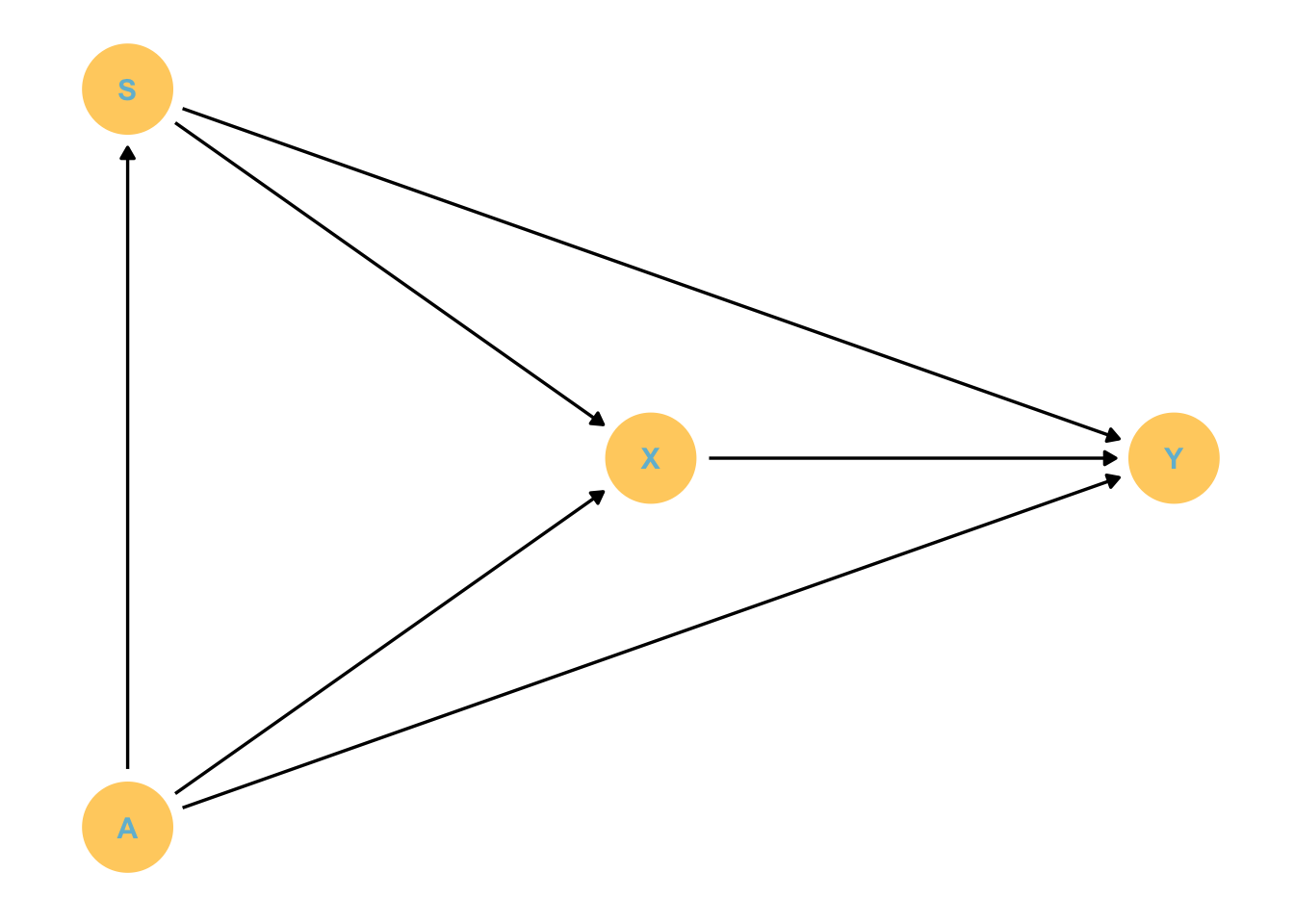
Code
dag_coords <-tibble(name =c("S", "A", "X", "Y"),x =c(1, 1, 2, 3),y =c(3, 1, 2, 2))dagify(S ~ A, X ~ S + A, Y ~ S + A + X,coords = dag_coords) %>%gg_simple_dag()
S = smoking
A = age
X = HIV
Y = stroke
We want to know the effect of X on Y - the effect of HIV on stroke risk.
Things to note:
Age has many effects, including some quite indirect ones
We need to decontaminate X of the effects of smoking and age.
Back-door criterion:
List paths
X -> Y
X <- S -> Y
X <- A -> Y
X <- A -> S -> Y
We need to close paths 2-4.
We can condition by A and S to effectively do this.
So we could fit a model that looks like this:
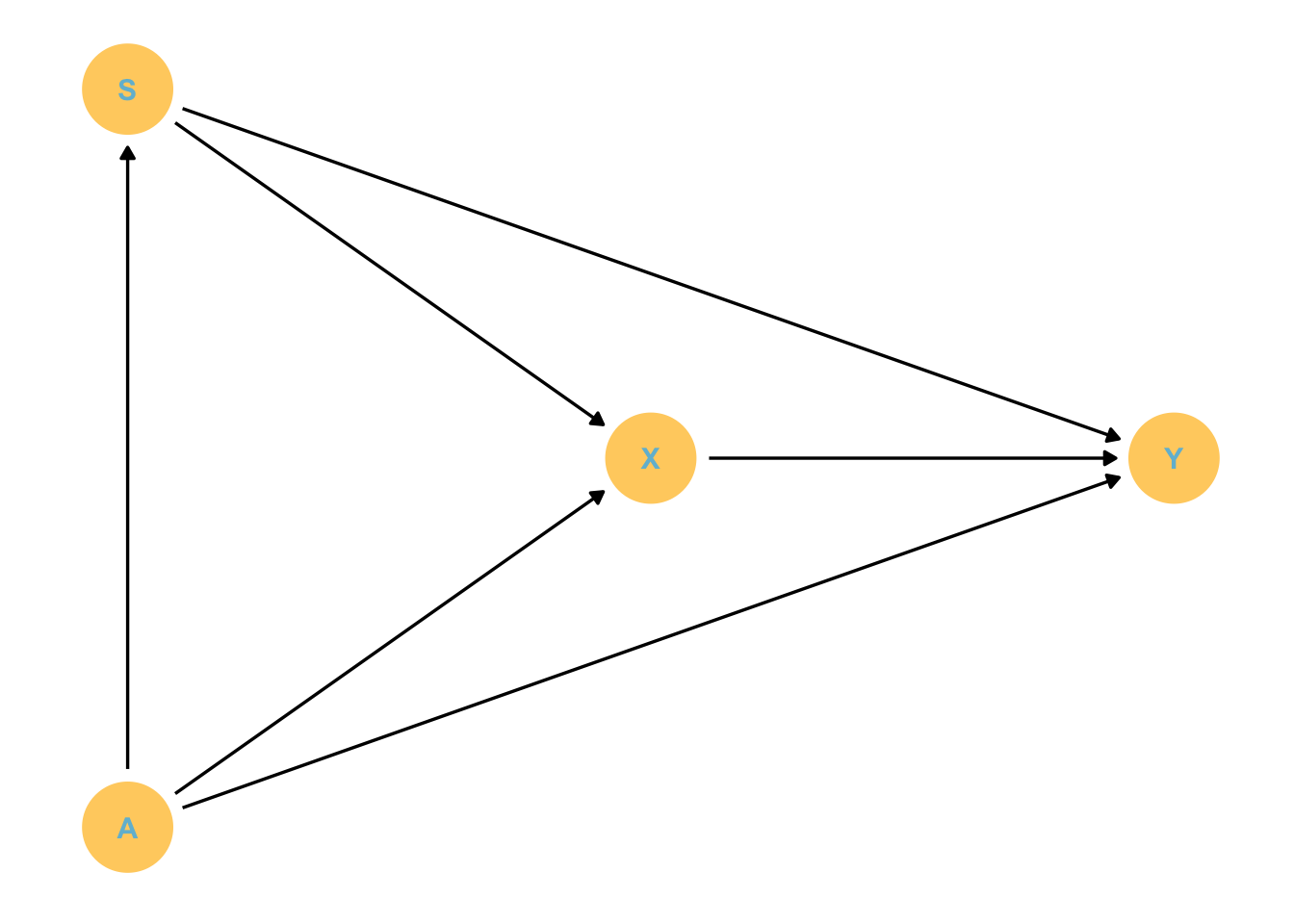
\[Y = X + S + A\] What if we focus on the effect of S?
Code
dagify(S ~ A, X ~ S + A, Y ~ S + A + X,coords = dag_coords) %>%gg_simple_dag()
S is confounded by A, as there is a back-door path. But we condition on A and X so that back-door path is closed.
BUT
What are we estimating for S? Well we have removed the indirect effect of S that acts through X, as we have closed that pipe. I.e. We are only estimating how smoking directly affects stroke, but not including how smoking acts on stroke through increasing the risk of HIV. This might be small or it might be large, we don’t know. The take home is this coefficient measures something different than that of X.
Now lets focus on Age.
It acts through everything, so we need not include any of the conditional variables to find its total effect on stroke (both direct and indirect). But we have conditioned on S and X. We have closed these pathways. We only get the direct effect, which may be tiny!
Take-home: don’t interpret all coefficients as unconditional effects!
How to report results
Either
Just report causal coefficients
Give an explicit interpretation of each coefficient
\(~\)
Lecture 7: Fitting Over and Under
\(~\)
Ockham’s razor: the simple solution is often the best. This lecture will focus on why this is.
We need to design efficient estimators: the struggle against data. Causual inference doesn’t help us much here.
Problems with prediction
To aid our understanding let’s use a Hominin dataset. We are interested in the affect of weight (mass) on brain volume.
Code
# create the data and print the tibble( hominin_data <-tibble(species =c("afarensis", "africanus", "habilis", "boisei", "rudolfensis", "ergaster", "sapiens"), brain =c(438, 452, 612, 521, 752, 871, 1350), mass =c(37.0, 35.5, 34.5, 41.5, 55.5, 61.0, 53.5)) )
# standardise. However, rescale the outcome, brain volume, so that the largest observed value is 1. Why not standardize brain volume as well? Because we want to preserve zero as a reference point: No brain at all. You can’t have negative brain. I don’t think.hominin_data <- hominin_data %>%mutate(mass_std = (mass -mean(mass)) /sd(mass),brain_std = brain /max(brain))# plot the relationship between brain volume and weighthominin_data %>%ggplot(aes(x = mass, y = brain, label = species)) +geom_point(size =5, colour =met.brewer("Cassatt1")[2]) +geom_text_repel(size =3, colour ="black", family ="Courier", seed =438, box.padding =1) +labs(subtitle ="Average brain volume by body\nmass for six hominin species",x ="Body mass (kg)",y ="Brain volume (cc)") +xlim(30, 65) +theme_classic() +theme(text =element_text(family ="Courier"),panel.background =element_rect(fill =alpha(met.brewer("Cassatt1", 8)[4], 1/4)))
What can we do with statistical models:
Find a function that describes these points (fitting)
What function explains these points (causal inference)
What happens when we change a point’s mass (intervention)
What is the next observation from the same process (forecasting or prediction)
We will focus on point 4 in this lecture.
\(~\)
Cross Validation
\(~\)
For simple models, more parameters improves fit within sample but may reduce accuracy out of sample
There is a trade off between flexibility and overfitting
We can test how effective our linear regression is at prediction using Leave-one-out-cross-validation
The steps:
Drop one point
Fit line to remaining points
Predict dropped point
Repeat (1) with next point
Score based on the error between actual point and predicted point of the regression
Code
hominin_brain_mass_model <-brm(data = hominin_data, family = gaussian, brain_std ~1+ mass_std,prior =c(prior(normal(0.5, 1), class = Intercept),prior(normal(0, 10), class = b),prior(lognormal(0, 1), class = sigma)),iter =2000, warmup =1000, chains =4, cores =4,seed =7,file ="fits/hominin_brain_mass_model")
Bayesian Cross-Validation
As usual we use distributions in bayes rather than points. The predictive distribution is uncertain, we don’t get a specific point estimate for the prediction. A cross-validation score is a bit harder to get over a distribution, but luckily we can follow the log pointwise predictive density equation. I won’t display it here because we can depend on the software. Just know that it is referred to as \(lppd_{CV}\). Essentially this goes point by point like above, finds the posterior distribution, then averages this distribution over the number of samples (I think).
Some definitions:
In sample score: summed deviance of points from the full sample regression
Out of sample score: summed deviance of points from each out of sample regression
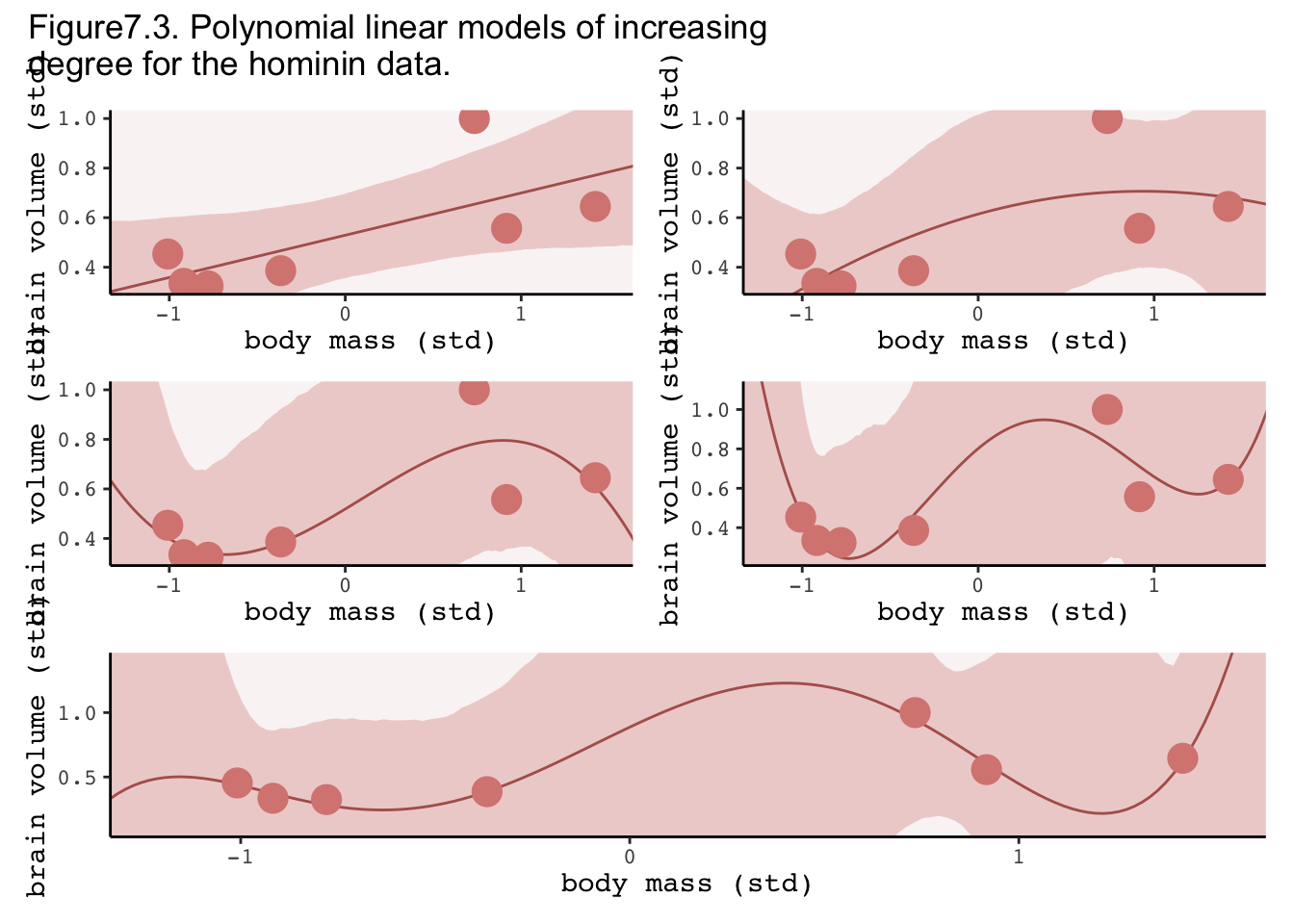
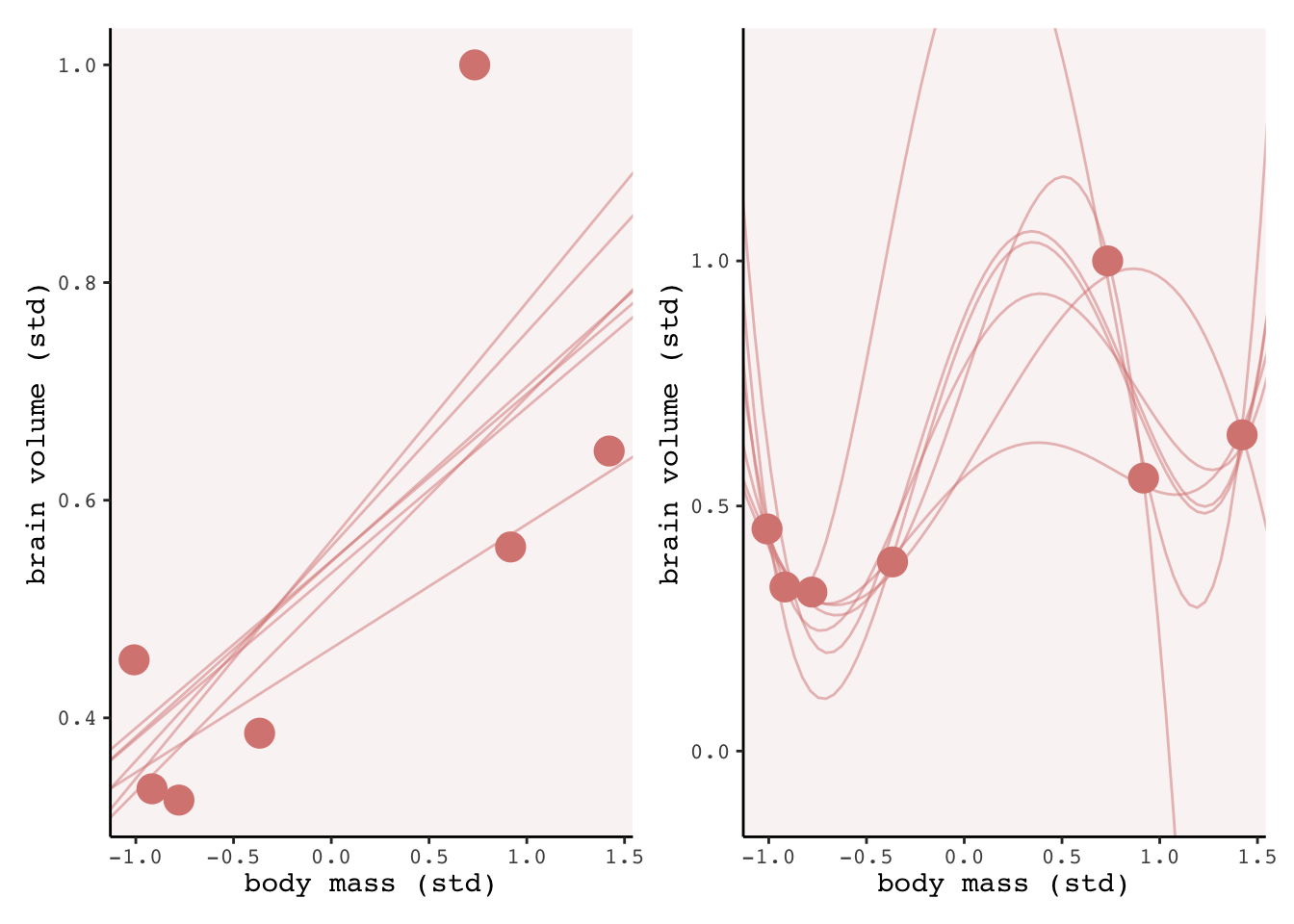
The in-sample score improves with model complexity, but the out of sample score gets worse (larger)! For the 4th order polynomial model in sample is insanely good, but the fit varies wildly once we remove a point. The take home is that we can build really complex models that fit the observed data really well, but have little to no predictive capacity for new data points. This is the basic trade-off inherent to modelling: over fitting leads to bad predictions.
But what if we have more data?
With more data we find that there is an intermediate model complexity (the 2nd degree model in this case) that maximises both within and out of sample deviance; that is, it is the best at describing these data. However, this doesn’t mean that it explains them, it’s just best at pure prediction in the absence of intervention. If we want to do this, we need to understand the causal structure.
From McElreath
The overfit polynomial models fit the data extremely well, but they suffer for this within-sample accuracy by making nonsensical out-of-sample predictions. In contrast, underfitting produces models that are inaccurate both within and out of sample. They learn too little, failing to recover regular features of the sample. (p. 201)
\(~\)
To explore the distinctions between overfitting and underfitting, we’ll need to refit the models above several times after serially dropping one of the rows in the data. You can filter() by row_number() to drop rows in a tidyverse kind of way. For example, we can drop the second row of hominin_data like this.
“Notice that the straight lines hardly vary, while the curves fly about wildly. This is a general contrast between underfit and overfit models: sensitivity to the exact composition of the sample used to fit the model” (p. 201).
Regularisation
\(~\)
We can design models that are better at prediction in their structure.
We want to identify regular features in the data, this leads to good predictions
We can minimise over fitting with:
Priors:
Sceptical priors have tighter variance, and reduce the flexibility of a model
Often tighter than we think they need to be
These improve predictions
Essentially, tight priors stop the model overreacting to nonsense within the sample
Choosing the width of a prior
For causal inference, use science, then go a touch tighter.
For pure prediction, we can use cross-validation to tune the prior.
Many tasks are a mix of both
Prediction penalty
For N points, cross-validation requires fitting N models. This quickly becomes computationally expensive, when your dataset gets larger.
Good news! We can compute the prediction penalty from a single model fit. There are two ways:
Importance sampling (PSIS)
Information criteria (WAIC or LOO)
\(~\)
Predictive criteria should not be casued to choose model structure
Fungus status is actually a better predictor of plant growth than anti-fungal treatment. Makes sense, the treatment is essentially an imperfect proxy for fungal status. For prediction this is better, but it’s terrible for determining the effect of the treatment. See that your preferred model depends entirely upon your aim.
Use a single posterior distribution for N points to sample for each posterior for N-1 points.
Key idea: Point with low probability has a strong influence on posterior distribution. Highly probable points on the other hand do not individually affect the posterior distribution much, because there are other points in close proximity conveying very similar information.
Can use pointwise probabilities to re-weight samples from the posterior.
BUT, importance sampling tends to be unreliable.
The best current one: Pareto-smoothed importance sampling
More stable (lower variance)
Useful diagnostics (it will tell you when it’s bad aka has high variance)
Identifies important (high leverage) points (outliers)
\(~\)
Information criteria
\(~\)
Akaike information criteria: AIC
The first form. For flat priors and large samples:
\(AIC = (-2) * lppd + 2k\)
Where -2 is a scaling term, lppd is the log pointwise predictive density and 2k is a penalty term for complexity, where k is the number of parameters.
This is a more complex formula but is has some similarities to the AIC formula.
\(\sum var_{\Theta} log p(y_{i}|\Theta)\) is the penalty term now.
\(WAIC\) provides a very similar result to PSIS, but without the diagnostics.
\(~\)
Outliers and robust regression
\(~\)
Some points are more important for the posterior distribution than others.
Outliers are observations in the tails of predictive distributions
If a model has outliers, this indicates the model doesn’t expect enough variation. This can lead to poor, damaging predictions.
Say we want to predict hurricane strength. We really don’t want to disregard extreme hurricanes, as this would be disastrous.
Don’t drop outliers, it’s the models fault, not the data’s. But how do we deal with them?
It’s the model that’s wrong, not the data
First, quantify influence of each point - we can do this with cross validation
Second, use a mixture model with fatter tails i.e. the student t distribution.
\(~\)
Robust regression
We’re back at the divorce rate example, where Idaho and Maine are outliers. Idaho has the lowest median marriage age and a very low divorce rate, while Maine has a very high divorce rate, but an average age of marriage.
Code
b5.3<-brm(data = Waffle_data, family = gaussian, d ~1+ m + a,prior =c(prior(normal(0, 0.2), class = Intercept),prior(normal(0, 0.5), class = b),prior(exponential(1), class = sigma)),iter =2000, warmup =1000, chains =4, cores =4,seed =5,file ="fits/b05.03")b5.3<-add_criterion(b5.3, criterion ="loo")b5.3<-add_criterion(b5.3, "waic", file ="fits/b05.03")Waffle_data %>%mutate(outlier =if_else(Location %in%c("Idaho", "Maine"), "YES", "NO")) %>%ggplot(aes(x = a, y = d, colour = outlier)) +geom_point(shape =1.5, stroke =2, size =3) +scale_colour_manual(values =c(met.brewer("Cassatt1")[2], met.brewer("Cassatt1")[6])) +geom_text_repel(aes(label = Location),data = Waffle_data %>%filter(Location ==c("Idaho", "Maine")), size =5, colour ="black", family ="Courier", seed =438, box.padding =0.5) +labs(x ="Age at marriage (std)",y ="Divorce rate (std)") +theme_classic() +theme(legend.position ="none")
Using PSIS we can find which data points are outliers
Code
loo(b5.3)
Computed from 4000 by 50 log-likelihood matrix
Estimate SE
elpd_loo -63.8 6.4
p_loo 4.7 1.9
looic 127.7 12.8
------
Monte Carlo SE of elpd_loo is 0.1.
Pareto k diagnostic values:
Count Pct. Min. n_eff
(-Inf, 0.5] (good) 49 98.0% 730
(0.5, 0.7] (ok) 1 2.0% 187
(0.7, 1] (bad) 0 0.0% <NA>
(1, Inf) (very bad) 0 0.0% <NA>
All Pareto k estimates are ok (k < 0.7).
See help('pareto-k-diagnostic') for details.
Ok so we have one point that is having a large effect on the posterior. Which is it?
Code
loo(b5.3) %>%pareto_k_ids(threshold =0.5)
[1] 13
Point 13, this means row 13 in the tibble. Lets find out which location that corresponds to.
Quadratic approximation (Laplace approximation, use McElreath’s quap function)
Markov chain Monte Carlo (intensive but more capable than anything else)
\(~\)
King Markov
\(~\)
King Markov rules over an archipelago with 8 islands. Each island has a different population size and the citizens yearn for him to visit. He splits his time between the islands like so:
He flips a coin to choose an island on the left or right of his current location
He then finds the population of the chosen island, or proposal island \(p_{2}\)
He then finds the population of his current island, \(p_{1}\)
He moves to the proposal island, with probability \(\frac{p_{2}}{p_{1}}\)
After a week he repeats from (1)
This procedure ensures visiting each island in proportion to its population, in the long run.
Knowing this, we can simulate and plot his journey:
Code
set.seed(1)num_weeks <-1e5positions <-rep(0, num_weeks)current <-10# we start at island 10for (i in1:num_weeks) {# record current position positions[i] <- current# flip coin to generate proposal proposal <- current +sample(c(-1, 1), size =1) # cool. sample 'flips the coin' so we either get -1 or 1, size specifies the number of coin flips# now make sure he loops around the archipelagoif (proposal <1) proposal <-10if (proposal >10) proposal <-1# does he move? prob_move <- proposal/current current <-ifelse(runif(1) < prob_move, proposal, current) # if prob_move is > 1 move to proposal}# Note that positions is now a numeric vector that has been populated by the simulationtibble(week =1:1e5,island = positions) %>%ggplot(aes(x = week, y = island)) +geom_point(shape =1, color ="salmon") +scale_x_continuous(breaks =seq(from =0, to =100, by =20)) +scale_y_continuous(breaks =seq(from =0, to =10, by =2)) +coord_cartesian(xlim =c(0, 100)) +labs(title ="Behold the Metropolis algorithm in action!",subtitle ="The dots show the king's path over the first 100 weeks.",x ="Week",y ="Island visited") +theme_bw() +theme(panel.grid =element_blank())
The number of weeks spent on each island is shown below, where island 10 has the largest population and island 1 the smallest. Note we have simmed for 1e5 weeks to show the tend in the long run.
Code
tibble(week =1:1e5,island = positions) %>%mutate(island =factor(island)) %>%ggplot(aes(x = island)) +geom_bar(fill ="salmon") +scale_y_continuous("number of weeks", expand =expansion(mult =c(0, 0.05))) +labs(title ="Old Metropolis shines in the long run.",subtitle ="Sure enough, the time the king spent on each island\nwas proportional to its population size.") +theme_bw() +theme(panel.grid =element_blank())
This is an example of Markov chain Monte Carlo
This is its simplest form: the metropolis algorithm
Islands = parameter values
Population size = posterior probability (how many ways can it explain the data)
Visit each parameter value in proportion to its posterior probability
We just need to find the posterior probability for two proposals at a time to build the distribution
\(~\)
Markov chains
\(~\)
Chain: a sequence of draws from distribution
Markov chain: history doesn’t matter, where you go next only matters on where you are now e.g. how the chain moves through parameter space
Monte Carlo: a numerical algorithm that uses randomisation to perform a calculation
- The King Markov example above follows the Metropolis algorithm.
- The Rosenbluth (Metropolis) was the first used in MCMC, introduced in 1953 (> 47,000 citations)
- We will use the Hamiltonian algorithm
The Rosenbluth algorithm
Moves are proposed, some are rejected and we progressively build a posterior by blindly stumbling around.
Think of parameter space like a bowl, it’s hard to climb the sides to unlikely posterior probabilities
The step size matters. That’s how far each proposal is from the current location.
A large step size is wasteful because many proposals are rejected, while a small step size is also poor as we don’t explore a large parameter space.
Code
# bowl simulation# this requires a lot of coding but we'll get there# first we need to simulate a bivariate distribution with a strong negative correlation# mean vectormu <-c(0, 0)# variance/covariance matrixsd_a1 <-0.22sd_a2 <-0.22rho <--.9Sigma <-matrix(data =c(sd_a1^2, rho * sd_a1 * sd_a2, rho * sd_a1 * sd_a2, sd_a2^2),nrow =2)# sample from the distribution with the `mvtnorm::rmvnorm()` functionset.seed(9)my_samples <- mvtnorm::rmvnorm(n =1000, mean = mu, sigma = Sigma)# We won’t actually be using the values from this simulation. Instead, we can evaluate the density function for this distribution using the mvtnorm::dmvnorm() function. But even before that, we’ll want to create a grid of values for the contour lines in Figure 9.3. Here we do so with a custom function called x_y_grid()# define the functionx_y_grid <-function(x_start =-1.6,x_stop =1.6,x_length =100,y_start =-1.6,y_stop =1.6,y_length =100) { x_domain <-seq(from = x_start, to = x_stop, length.out = x_length) y_domain <-seq(from = y_start, to = y_stop, length.out = y_length) x_y_grid_tibble <- tidyr::expand_grid(a1 = x_domain, a2 = y_domain)return(x_y_grid_tibble)}# simulatecontour_plot_dat <-x_y_grid()# now compute density values for each combination of a1 and a2contour_plot_dat <- contour_plot_dat %>%mutate(d = mvtnorm::dmvnorm(as.matrix(contour_plot_dat), mean = mu, sigma = Sigma))# now we can create our base contour plotcontour_plot <- contour_plot_dat %>%ggplot() +geom_contour(aes(x = a1, y = a2, z = d), size =1/8, color ="#FFB300",breaks =9^(-(10*1:25))) +scale_x_continuous(expand =c(0, 0)) +scale_y_continuous(expand =c(0, 0)) +theme_bw() +theme(panel.grid =element_blank(),panel.background =element_rect(fill ="#FFF9C4"))# Now define the metropolis/rosenbluth algorithmmetropolis <-function(num_proposals =50,step_size =0.1,starting_point =c(-1, 1)) {# Initialize vectors where we will keep track of relevant candidate_x_history <-rep(-Inf, num_proposals) candidate_y_history <-rep(-Inf, num_proposals) did_move_history <-rep(FALSE, num_proposals)# Prepare to begin the algorithm... current_point <- starting_pointfor(i in1:num_proposals) {# "Proposals are generated by adding random Gaussian noise# to each parameter" noise <-rnorm(n =2, mean =0, sd = step_size) candidate_point <- current_point + noise# store coordinates of the proposal point candidate_x_history[i] <- candidate_point[1] candidate_y_history[i] <- candidate_point[2]# evaluate the density of our posterior at the proposal point candidate_prob <- mvtnorm::dmvnorm(candidate_point, mean = mu, sigma = Sigma)# evaluate the density of our posterior at the current point current_prob <- mvtnorm::dmvnorm(current_point, mean = mu, sigma = Sigma)# Decide whether or not we should move to the candidate point acceptance_ratio <- candidate_prob / current_prob should_move <-ifelse(runif(n =1) < acceptance_ratio, TRUE, FALSE)# Keep track of the decision did_move_history[i] <- should_move# Move if necessaryif(should_move) { current_point <- candidate_point } }# once the loop is complete, store the relevant results in a tibble results <- tibble::tibble(candidate_x = candidate_x_history,candidate_y = candidate_y_history,accept = did_move_history )# compute the "acceptance rate" by dividing the total number of "moves"# by the total number of proposals number_of_moves <- results %>% dplyr::pull(accept) %>%sum(.) acceptance_rate <- number_of_moves/num_proposalsreturn(list(results = results, acceptance_rate = acceptance_rate))}# run the algorithm with step size = 0.1 to create the data for the left figureset.seed(9)round_1 <-metropolis(num_proposals =50,step_size =0.1,starting_point =c(-1,1))# create the figurep1 <- contour_plot +geom_point(data = round_1$results,aes(x = candidate_x, y = candidate_y, color = accept, shape = accept)) +labs(subtitle =str_c("step size 0.1,\naccept rate ", round_1$acceptance_rate),x ="a1",y ="a2") +scale_shape_manual(values =c(21, 19)) +scale_color_manual(values =c("black", "#8D6E63"))# now run the algorithm with step size 0.25# simulateset.seed(9)round_2 <-metropolis(num_proposals =50,step_size =0.25,starting_point =c(-1,1))# plotp2 <- contour_plot +geom_point(data = round_2$results,mapping =aes(x = candidate_x, y = candidate_y, color = accept, shape = accept)) +scale_shape_manual(values =c(21, 19)) +scale_color_manual(values =c("black", "#8D6E63")) +scale_y_continuous(NULL, breaks =NULL, expand =c(0, 0)) +labs(subtitle =str_c("step size 0.25,\naccept rate ", round_2$acceptance_rate),x ="a1")# combine(p1 + p2) +plot_annotation(title ="Metropolis chains under high correlation") +plot_layout(guides ="collect")
\(~\)
Hamiltonian Monte Carlo
\(~\)
This is like letting a skateboard roll in a bowl and tracking where it stops.
Then from that point for flick the board through the bowl again and repeat.
Once again step size matters
Long steps are susceptible to U-turns which makes sampling inefficient
When posteriors get complicated, algorithms like the Rosenbluth struggle to find proposals that aren’t rejected, meaning they can’t move through parameter space. They get stuck. Hamiltonian Monte Carlo does not share this struggle.
Ok, but our computer needs to know the curvature of the parameter space in order to effectively ‘flick the skateboard’. More formally, we need gradients. How can we program this?
We certainly don’t want to write them for every point in the parameter space
Auto-diff (automatic differentiation) saves the day
How this works is a bit beyond me, but the computer knows how (Stan <3)
\(~\)
Analysing the data: Divorce dataset
\(~\)
Note that McElreath introduces the Wines2012 dataset in his lecture, but I’m lazy so I use a model we’ve already fit.
We already have been using Hamiltonian Monte Carlo to fit our brms models. So nothing really new here for us. In fact we can just reload our previously fitted Marriage model from Lecture 5.
Family: gaussian
Links: mu = identity; sigma = identity
Formula: d ~ 1 + a
Data: Waffle_data (Number of observations: 50)
Draws: 4 chains, each with iter = 2000; warmup = 1000; thin = 1;
total post-warmup draws = 4000
Population-Level Effects:
Estimate Est.Error l-95% CI u-95% CI Rhat Bulk_ESS Tail_ESS
Intercept 0.00 0.10 -0.20 0.20 1.00 3537 2678
a -0.57 0.12 -0.79 -0.34 1.00 3801 2753
Family Specific Parameters:
Estimate Est.Error l-95% CI u-95% CI Rhat Bulk_ESS Tail_ESS
sigma 0.82 0.09 0.68 1.01 1.00 2735 2675
Draws were sampled using sampling(NUTS). For each parameter, Bulk_ESS
and Tail_ESS are effective sample size measures, and Rhat is the potential
scale reduction factor on split chains (at convergence, Rhat = 1).
Note in our summary output we have a number of diagnostics.
\(~\)
Diagnostics
\(~\)
Trace plots
Code
post <-posterior_samples(Marriage_age_model, add_chain = T)mcmc_trace(post[, c(1:3, 8)], # we need to include column 7 because it contains the chain info facet_args =list(ncol =1), size = .15) +labs(title ="My custom trace plots") +theme_minimal() +theme(panel.grid =element_blank())
These plot a set number of samples from the Markov chain for each parameter
Warm-up:
Early chains where stan is figuring out step size and how long the trajectory should be.
Warmup samples are not representative of the target posterior distribution, no matter how long warmup continues. They are not burning in, but rather more like cycling the motor to heat things up and get ready for sampling. When real sampling begins, the samples will be immediately from the target distribution, assuming adaptation was successful.
We don’t use it for estimation, and it is not shown in our plot above.
Good trace plots look stationary. That means they are centered around an area of high probability i.e. the lower part of the half-pipe
It is important to run more than 1 chain to check for convergence.
Convergence: each chain explores the right distribution and every chain explores the same distribution
Start them at different points in parameter space and they should converge on the same posterior
4 chains is generally good
We can also plot the chains with plot
Code
plot(Marriage_age_model)
\(~\)
Trace rank plots (Trank plots)
x axis is sequential samples, like a trace plot
y axis shows rank orders across the chains
Quick visual way to check if one of the chains is consistently above or below all of the others
Ideally the chain on top should be constantly switching
Start and end of each chain to explore the same region e.g. consistency across samples
Independent chains to explore the same region e.g. consistency across chains
R-hat: A ratio of variances
As total variance among all chains shrinks to average variance within chains, R-hat approaches 1
All variance should ideally be within chains not between them if they converge on the same posterior distribution
1 is good, but it also doesn’t ensure your sampling is good
\(~\)
n_eff
The effective number of samples
Put another way “How long would the chain be, if each sample was independent of the one before it?”
Because samples are autocorrelated, that is those in close sequential order are often exploring a similar parameter space, they convey less information than would a truly independent sampling approach.
The greater this autocorrelation, the fewer effective samples you have.
This doesn’t mean it’s bad, it’s just inefficient and you’ll have to sample for longer.
n_eff will nearly always be smaller than the specified number of samples because of autocorrelation.
brms returns two values: Bulk_ESS and Tail_ESS
Bulk_ESS = n_eff
Tail_ESS = n_eff at the extremes of intervals
Code
post %>%mcmc_acf(pars =vars(b_Intercept:sigma),lags =5) +theme(panel.grid =element_blank())
This is a healthy autocorrelation plot
\(~\)
Divergent transitions
This is a kind of rejected proposal
If the posterior distribution has a very steep gradient it can lead to many rejected proposals, even for HMC
They aren’t bad in isolation, Rosenbluth algorithm has loads
Lots of divergent transitions mean that the algorithm is exploring the posterior distribution very poorly and that it might be stuck in one spot and missing some of the parameter space
\(~\)
Bad chains
\(~\)
Lets fit an awful model. Give it two data points and stupidly flat priors.
Code
b9.2<-brm(data =list(y =c(-1, 1)), family = gaussian, y ~1,prior =c(prior(normal(0, 1000), class = Intercept),prior(exponential(0.0001), class = sigma)),iter =2000, warmup =1000, chains =3,seed =9,file ="fits/b09.02")b9.2
Family: gaussian
Links: mu = identity; sigma = identity
Formula: y ~ 1
Data: list(y = c(-1, 1)) (Number of observations: 2)
Draws: 3 chains, each with iter = 2000; warmup = 1000; thin = 1;
total post-warmup draws = 3000
Population-Level Effects:
Estimate Est.Error l-95% CI u-95% CI Rhat Bulk_ESS Tail_ESS
Intercept 0.37 310.88 -691.22 683.09 1.08 472 385
Family Specific Parameters:
Estimate Est.Error l-95% CI u-95% CI Rhat Bulk_ESS Tail_ESS
sigma 515.45 1217.56 13.14 3568.10 1.05 44 78
Draws were sampled using sampling(NUTS). For each parameter, Bulk_ESS
and Tail_ESS are effective sample size measures, and Rhat is the potential
scale reduction factor on split chains (at convergence, Rhat = 1).
The trace plot
Code
post <-posterior_samples(b9.2, add_chain = T)p1 <- post %>%mcmc_trace(pars =vars(b_Intercept:sigma),size = .25)p2 <- post %>%mcmc_rank_overlay(pars =vars(b_Intercept:sigma))( (p1 / p2) &scale_color_manual(values =c("#7CB342", "#FDD835", "#FB8C00", "#F4511E")) &theme(legend.position ="none")) +plot_annotation(subtitle ="These chains are not healthy")
Very bad.
The problem with this model is the priors. Can we fix it with better priors?
Code
b9.3<-brm(data =list(y =c(-1, 1)), family = gaussian, y ~1,prior =c(prior(normal(1, 10), class = Intercept),prior(exponential(1), class = sigma)),iter =2000, warmup =1000, chains =3,seed =9,file ="fits/b09.03")b9.3
Family: gaussian
Links: mu = identity; sigma = identity
Formula: y ~ 1
Data: list(y = c(-1, 1)) (Number of observations: 2)
Draws: 3 chains, each with iter = 2000; warmup = 1000; thin = 1;
total post-warmup draws = 3000
Population-Level Effects:
Estimate Est.Error l-95% CI u-95% CI Rhat Bulk_ESS Tail_ESS
Intercept 0.03 1.13 -2.21 2.50 1.00 945 624
Family Specific Parameters:
Estimate Est.Error l-95% CI u-95% CI Rhat Bulk_ESS Tail_ESS
sigma 1.54 0.84 0.60 3.71 1.00 909 1245
Draws were sampled using sampling(NUTS). For each parameter, Bulk_ESS
and Tail_ESS are effective sample size measures, and Rhat is the potential
scale reduction factor on split chains (at convergence, Rhat = 1).
The trace plot
Code
post <-posterior_samples(b9.3, add_chain = T)p1 <- post %>%mcmc_trace(pars =vars(b_Intercept:sigma),size = .25)p2 <- post %>%mcmc_rank_overlay(pars =vars(b_Intercept:sigma))( (p1 / p2) &scale_color_manual(values =c("#7CB342", "#FDD835", "#FB8C00", "#F4511E")) &theme(legend.position ="none")) +plot_annotation(subtitle ="Weakly informative priors to the rescue")
Note that sigma is upwardly distributed. This is normal for a scale parameter.
\(~\)
Lecture 9: Modelling events
\(~\)
Modelling events
Events: discrete, unordered outcomes
Observations are counts
Unknowns are probabilities, or odds (transformed probabilities)
Odds are probs event happens / prob it does not
Everything interacts always everywhere!
A beast known as ‘log-odds’ - the logarithm of the odds.
\(~\)
UC Berkeley admissions
\(~\)
4526 graduate school applications for 1973 UC Berkeley. The data is stratified by department and gender.
# A tibble: 12 × 5
dept applicant.gender admit reject applications
<fct> <fct> <int> <int> <int>
1 A male 512 313 825
2 A female 89 19 108
3 B male 353 207 560
4 B female 17 8 25
5 C male 120 205 325
6 C female 202 391 593
7 D male 138 279 417
8 D female 131 244 375
9 E male 53 138 191
10 E female 94 299 393
11 F male 22 351 373
12 F female 24 317 341
Right, let’s start with the OWL infrastructure.
and 2) together. Find the estimand and describe the scientific model
D = Department e.g. science, arts etc - some departments have high competitive admission, whereas others have low, where it’s easier to achieve admission.
A = Admission
Department is a mediator. This is a very common causal structure.
What do we mean by the causal effect of gender?
How does the referee perceive someone based on gender. This can be blinded, at least to some extent.
But gender also affects where someone applies…
Which of the paths in this DAG are discrimination?
The direct path is status-based or taste-based (refs are biased for or against individuals of particular genders)
Indirect path is structural discrimination e.g. unequal access to higher education by greater funding for a gender biased program
Total: both paths: this is what people experience
Build the model with some simulated data
Code
# generative model, basic mediator scenarioN <-1000# number of applicationsG <-sample(1:2, size = N, replace = T) # close to even gender distribution, like flipping a coin N timesD <-rbernoulli(N, if_else(G ==1, 0.3, 0.8)) +1# gender 1 individuals tend to apply to department 1, G2 applies to d 2 more often# matrix of acceptance ratesaccept_rate <-matrix(c(0.1, 0.3, 0.1, 0.3), nrow =2) # rows are departments, columns are genders. Note we code no discrimination# simulate acceptanceA <-rethinking::rbern(N, accept_rate[D, G])table(G, D)
D
G 1 2
1 349 136
2 109 406
The Table illustrates that Gender 1 applies to department 1 more often than does gender 2.
Code
table(G, A)
A
G 0 1
1 401 84
2 385 130
This Table shows that Gender 2 gets accepted more often than Gender 1. This is because it’s easier to get into department 2. This is the mediating path.
We get the same pattern! This means that we cannot tell which causal path is dictating the effect. So we must turn to the backdoor criteria.
\(~\)
Generalised Linear Models
\(~\)
A linear model is part of a larger family of models called generalised linear models. Generalised linear models have an expected value that is some function of an additive combination of parameters (linear models are this but without the function).
The linear model can take any real value, but if we have a bernoulli data generating process we need to limit these values within a range of 0 and 1.
Distributions:
The relative number of ways to observe data, given assumptions
Distributions are matched to constraints on the observed data e.g. counts can’t be negative
Link functions match distributions (brms will give you these)
The notation:
\(Y_{i} = Bernoulli(p_i)\)
\(f(p_i) = \alpha + \beta_XX_i + B_ZZ_i\)
\(Y_i\) is a 0/1 outcome
\(p_i\) is the probability of the event
\(\alpha + \beta_XX_i + B_ZZ_i\) is identical to the linear model and can take any real value
\(f()\)maps the probability scale to the linear scale
\(~\)
Link functions
\(f\) is the link function.
Links parameters of distribution to linear model
Code
tibble(x =seq(from =-1, to =3, by = .01)) %>%mutate(probability = .35+ x * .5) %>%ggplot(aes(x = x, y = probability)) +geom_rect(xmin =-1, xmax =3,ymin =0, ymax =1,fill =met.brewer("Hiroshige")[6]) +geom_hline(yintercept =0:1, linetype =2, color =met.brewer("Hiroshige")[5]) +geom_line(aes(linetype = probability >1, color = probability >1),size =1) +geom_segment(x =1.3, xend =3,y =1, yend =1,size =2/3, color =met.brewer("Hiroshige")[3]) +annotate(geom ="text",x =1.28, y =1.04, hjust =1,label ="This is why we need link functions",color =met.brewer("Hiroshige")[4], size =2.6) +scale_color_manual(values =c(met.brewer("Hiroshige")[3:4])) +scale_y_continuous(breaks =c(0, .5, 1)) +coord_cartesian(xlim =c(0, 2),ylim =c(0, 1.2)) +theme(legend.position ="none",panel.background =element_rect(fill =met.brewer("Hiroshige")[7]),panel.grid =element_blank())
\(f^{-1}\) is the inverse link function. The purpose of this is to undo the effect of the link function.
What the computer is actually doing is this:
\(p_i = f^{-1}(\alpha + \beta_XX_i + B_ZZ_i)\)
It undoes the transformation
\(~\)
The logit link
Bernoulli/binomial models often use the logit link.
This is the log-odds of the event happening.
\(logit(p_i) = log \frac{p_i}{1 - p_i}\)
\(\frac{p_i}{1 - p_i}\) is the odds
Why do we take the log of the odds? It is the right transform to convert the real number line to the probability interval 0-1 without causing problems.
This wording helps explain why we use the inverse function
The logit function makes S curves or logistic curves. This is because they must bend and flatten at 0 and 1
Code
# first, we'll make data for the horizontal linesalpha <-0beta <-4lines <-tibble(x =seq(from =-1, to =1, by = .25)) %>%mutate(`log-odds`= alpha + x * beta,probability =exp(alpha + x * beta) / (1+exp(alpha + x * beta)))# now we're ready to make the primary databeta <-2d <-tibble(x =seq(from =-1.5, to =1.5, length.out =50)) %>%mutate(`log-odds`= alpha + x * beta,probability =exp(alpha + x * beta) / (1+exp(alpha + x * beta))) # now we make the individual plotsp1 <- d %>%ggplot(aes(x = x, y =`log-odds`)) +geom_hline(data = lines,aes(yintercept =`log-odds`),color =met.brewer("Hiroshige")[2]) +geom_line(size =1.5, color =met.brewer("Hiroshige")[7]) +coord_cartesian(xlim =c(-1, 1)) +theme(panel.background =element_rect(fill =met.brewer("Hiroshige")[5]),panel.grid =element_blank())p2 <- d %>%ggplot(aes(x = x, y = probability)) +geom_hline(data = lines,aes(yintercept = probability),color =met.brewer("Hiroshige")[2]) +geom_line(size =1.5, color =met.brewer("Hiroshige")[7]) +coord_cartesian(xlim =c(-1, 1)) +theme(panel.background =element_rect(fill =met.brewer("Hiroshige")[5]),panel.grid =element_blank())# finally, we're ready to mash the plots together and behold their nerdy glory(p1 | p2) +plot_annotation(subtitle ="The logit link transforms a linear model (left) into a probability (right).")
How to read the log-odds scale:
logit(p) = 0, p = 0.5 (happens half the time)
logit(p) = 4, p = nearly always happens (very close to 1 odds)
logit(p) = -4, p = nearly never happens (very close to 0 odds)
\(~\)
Setting priors:
We need to know what happens when we convert the prior distribution to the probability scale
For , if we set wide priors we make the model assume that the event will either never or always happen
If we set a prior of ~ Normal(0, 1) we start to get some skepticism of extreme values but not much! - See plot below
For : to not assume very strong relationships we need something like: ~ Normal(0, 0.5)
\(~\)
Lets look at how bad even Normal(0, 2) for an intercept prior is:
Code
hist(rnorm(1000, 0, 2))
Code
data <-rnorm(1000, 0, 2) %>% as_tibble %>%mutate(prob_space =inv_logit_scaled(value)) p1 <- data %>%ggplot(aes(x = value)) +geom_histogram() +xlab("log-odds")p2 <- data %>%ggplot(aes(x = prob_space)) +geom_histogram() +xlab("probability")p1 + p2
\(~\)
Analysing UC Berkelely data
\(~\)
Look at the data again
Code
UCB_admissions
# A tibble: 12 × 5
dept applicant.gender admit reject applications
<fct> <fct> <int> <int> <int>
1 A male 512 313 825
2 A female 89 19 108
3 B male 353 207 560
4 B female 17 8 25
5 C male 120 205 325
6 C female 202 391 593
7 D male 138 279 417
8 D female 131 244 375
9 E male 53 138 191
10 E female 94 299 393
11 F male 22 351 373
12 F female 24 317 341
First we should talk about the difference between Bernoulli and Binomial regression.
They are interchangeable from an inference perspective
Bernoulli data is displayed as 0, 1 data, while binomial variables are aggregated counts of a bernoulli process.
Note that the data above is aggregated, so we should specify a binomial distribution in our upcoming model
First let’s get the total effect of gender on admission acceptance
Family: binomial
Links: mu = logit
Formula: admit | trials(applications) ~ 0 + applicant.gender
Data: UCB_admissions (Number of observations: 12)
Draws: 3 chains, each with iter = 2000; warmup = 1000; thin = 1;
total post-warmup draws = 3000
Population-Level Effects:
Estimate Est.Error l-95% CI u-95% CI Rhat Bulk_ESS
applicant.genderfemale -0.83 0.05 -0.93 -0.73 1.00 2481
applicant.gendermale -0.22 0.04 -0.29 -0.14 1.00 2387
Tail_ESS
applicant.genderfemale 2063
applicant.gendermale 1968
Draws were sampled using sampling(NUTS). For each parameter, Bulk_ESS
and Tail_ESS are effective sample size measures, and Rhat is the potential
scale reduction factor on split chains (at convergence, Rhat = 1).
Output tables suck when we have lots of parameters. Contrasts between treatment levels are far more informative.
Let’s find the difference in admissions between genders
Code
post <-as_draws_df(UCB_admissions_model) %>%mutate(f_mean =inv_logit_scaled(b_applicant.genderfemale),m_mean =inv_logit_scaled(b_applicant.gendermale),diff = f_mean - m_mean)post %>%# plotggplot(aes(x = diff)) +geom_vline(xintercept =0, linetype =2) +stat_halfeye(point_interval = median_qi, .width = .95,color ="black",fill =met.brewer("Hiroshige")[4]) +labs(x =" Mean effect of being female on application acceptance",y =NULL) +coord_cartesian(xlim =c(-0.25, 0)) +theme(axis.ticks.y =element_blank(),legend.position ="none") +theme_tidybayes()
There is one department where women are advantaged over men, but none where men and advantaged. The 14% penalty found in the total effect has disappeared
\(~\)
Survival analysis bonus
\(~\)
Survival analysis is a form of count model. Once again the parameters are about rates.
What to do with data that wasn’t counted? i.e. you have some data you were collecting that did not reach the end point e.g. a cat escaped before it was adopted, a fly line was undone by a mistake rather than an extinction event.
We can tackle this with censoring:
Left censoring: don’t know when time started
Right censoring: something cut observation off before the event occurred
Censoring matters because we bias our data by dropping the data e.g. imagine estimating PhD time to completion but leaving out all the drop-outs
Time to event (out-event) - most cats were adopted
Event either 1 (adopted) or 2 (something else - death, escape, censored - the cat is still not adopted at time of data download)
Cat colour is also known
Data distributions we can use:
Exponential:
From McElreath
“It is a fundamental distribution of distance and duration, kinds of measurements that represent displacement from some point of reference, either in time or space. If the probability of an event is constant in time or across space, then the distribution of events tends towards exponential. The exponential distribution has maximum entropy among all non-negative continuous distributions with the same average displacement.” (p. 314).
The rate of decay is constant e.g. like nuclear decay with half-lives
Log-link
\(f(y) = \lambda e^{-\lambda y}\)
Where y is a non-negative continuous variable and is called the rate.
Importantly, the mean of the exponential distribution is the inverse of the rate:
\(E[y] = \frac{1}{\lambda}\)
This is important because it is how brms parameterises exponential models.
How are the two events distributed i.e. adoption versus censor
Code
cat_data %>%mutate(censored =factor(censored)) %>%filter(days_to_event <300) %>%ggplot(aes(x = days_to_event, y = censored)) +# let's just mark off the 50% intervalsstat_halfeye(.width = .5, fill =met.brewer("Hokusai2")[2], height =4) +scale_y_discrete(NULL, labels =c("censored == 0", "censored == 1")) +coord_cartesian(ylim =c(1.5, 5.1)) +theme(axis.ticks.y =element_blank()) +theme_tidybayes()
Do note there is a very long right tail that we’ve cut off for the sake of the plot. Anyway, the point of this plot is to show that the distribution for our primary variable, days_to_event, looks very different conditional on whether the data were censored. As McElreath covered in the lecture, we definitely don’t want to lose that information by excluding the censored cases from the analysis.
Family: exponential
Links: mu = log
Formula: days_to_event | cens(censored) ~ 0 + black
Data: cat_data (Number of observations: 22356)
Draws: 4 chains, each with iter = 2000; warmup = 1000; thin = 1;
total post-warmup draws = 4000
Population-Level Effects:
Estimate Est.Error l-95% CI u-95% CI Rhat Bulk_ESS Tail_ESS
blackblack 4.05 0.03 4.00 4.10 1.00 3122 2128
blackother 3.88 0.01 3.86 3.90 1.00 2925 2466
Draws were sampled using sampling(NUTS). For each parameter, Bulk_ESS
and Tail_ESS are effective sample size measures, and Rhat is the potential
scale reduction factor on split chains (at convergence, Rhat = 1).
Since we modelled \(log \mu_{i}\), we need to transform our parameters back into the metric using the formula:
# annotationtext <-tibble(color =c("black", "other"),days =c(40, 34),p =c(.55, .45),label =c("black cats", "other cats"),hjust =c(0, 1))# wranglef <-fixef(b11.15) %>%data.frame() %>%rownames_to_column() %>%mutate(color =str_remove(rowname, "black")) %>%# most important step. We predict over 100 days, assuming a constant rate of declineexpand(nesting(Estimate, Q2.5, Q97.5, color), days =0:100) %>%mutate(m =1-pexp(days, rate =1/exp(Estimate)),ll =1-pexp(days, rate =1/exp(Q2.5)),ul =1-pexp(days, rate =1/exp(Q97.5)))# plot!f %>%ggplot(aes(x = days)) +geom_hline(yintercept = .5, linetype =3, color =met.brewer("Hokusai2")[2]) +geom_ribbon(aes(ymin = ll, ymax = ul, fill = color),alpha =1/2) +geom_line(aes(y = m, color = color)) +geom_text(data = text,aes(y = p, label = label, hjust = hjust, color = color),family ="Times") +scale_fill_manual(values =met.brewer("Hokusai2")[c(4, 1)], breaks =NULL) +scale_color_manual(values =met.brewer("Hokusai2")[c(4, 1)], breaks =NULL) +scale_y_continuous("proportion remaining", breaks =c(0, .5, 1), limits =0:1) +xlab("days to adoption")
Another way to explore this model is to ask: About how many days would it take for half of the cats of a given colour to be adopted? We can do this with help from the qexp() function. For example:
The model suggests it takes about six days longer for the half of the black cats to be adopted.
\(~\)
Lecture 10: Counts and Confounds
\(~\)
Linear models: expected value is additive (linear) combination of parameters. What you see is what you get.
Generalised linear models: these are like tide prediction models. They have many gears that are difficult to interpret. More formally, the expected value is some function of an additive combination of parameters. If we leave this expected value on the function scale, it means very little to the common man. We must strive to show the ‘tide prediction’.
It is very important to note that uniform changes in the predictor do not lead to uniform changes in prediction. I.e. think of how values change on the log-odds scale.
All predictor variables interact - if one predictor pushes the prediction towards 0 or 1 then the effect of other predictors will be smaller.
\(~\)
UC Berkeley continued - Confounded admissions
\(~\)
Code
dag_coords <-tibble(name =c("G", "D", "A", "u"),x =c(1, 2, 3, 3.2),y =c(1, 2, 1, 2.2))dagify(A ~ G + D + u, D ~ G + u,coords = dag_coords) %>%gg_simple_dag()
What if \(u\), the students ability affects both department choice and acceptance rate. We do not have this in our dataset, but it still may be present in our scientific model.
Let’s simulate such a situation. Here are some general tips for simulation:
Simulate variables with no parents (they don’t depend on other variables)
Simulate the values that require parents
Code
set.seed(17)N <-2000# number of applicants# even gender distributionG <-sample(1:2, size = N, replace =TRUE)# sample ability, high (1) to average (0)u <- rethinking::rbern(N, 0.1) # gender 1 tends to apply to department 1, 2 to 2# also G =1 with greater ability tend to apply to 2 as wellD <- rethinking::rbern(N, if_else(G ==1, u *1, 0.75)) +1# individuals of high ability have a 50% chance of applying for department 2# matrix of acceptance ratesaccept_rate_u0 <-matrix(c(0.1, 0.1, 0.1, 0.3), nrow =2) # average ability, with discrimintation in dep 2accept_rate_u1 <-matrix(c(0.3, 0.3, 0.5, 0.5), nrow =2) # high ability# simulate acceptanceP <-sapply(1:N, function(i)if_else(u[i] ==0, accept_rate_u0[D[i], G[i]], accept_rate_u1[D[i], G[i]] ))A <- rethinking::rbern(N, P)data_sim <-tibble(Accepted = A, Department =as.factor(D), Gender =as.factor(G))
Family: bernoulli
Links: mu = logit
Formula: Accepted ~ 0 + Gender
Data: data_sim (Number of observations: 2000)
Draws: 3 chains, each with iter = 2000; warmup = 1000; thin = 1;
total post-warmup draws = 3000
Population-Level Effects:
Estimate Est.Error l-95% CI u-95% CI Rhat Bulk_ESS Tail_ESS
Gender1 -1.91 0.09 -2.09 -1.74 1.01 2500 1946
Gender2 -1.09 0.07 -1.23 -0.95 1.00 2746 1836
Draws were sampled using sampling(NUTS). For each parameter, Bulk_ESS
and Tail_ESS are effective sample size measures, and Rhat is the potential
scale reduction factor on split chains (at convergence, Rhat = 1).
This model gets the total effect of gender correct. Gender 1 is disadvantaged overall.
The direct effect of gender
Code
UCB_admissions_model_ability_direct <-brm(data = data_sim, family = bernoulli, Accepted ~1+ Gender * Department,prior =c(prior(normal(0, 1), class = Intercept),prior(normal(0, 1), class = b)),iter =2000, warmup =1000, cores =3, chains =3,seed =11,file ="fits/UCB_admissions_model_ability_direct")UCB_admissions_model_ability_direct
Family: bernoulli
Links: mu = logit
Formula: Accepted ~ 1 + Gender * Department
Data: data_sim (Number of observations: 2000)
Draws: 3 chains, each with iter = 2000; warmup = 1000; thin = 1;
total post-warmup draws = 3000
Population-Level Effects:
Estimate Est.Error l-95% CI u-95% CI Rhat Bulk_ESS Tail_ESS
Intercept -2.12 0.11 -2.33 -1.92 1.00 2287 2096
Gender2 -0.28 0.24 -0.76 0.17 1.00 1786 1653
Department2 1.24 0.23 0.79 1.70 1.00 1881 1772
Gender2:Department2 0.35 0.32 -0.27 0.98 1.00 1529 1668
Draws were sampled using sampling(NUTS). For each parameter, Bulk_ESS
and Tail_ESS are effective sample size measures, and Rhat is the potential
scale reduction factor on split chains (at convergence, Rhat = 1).
Model 2 is confounded. To show this we should look at the contrasts - don’t look at the gears!
This means we can’t estimate the direct effect of D or G
What’s happening here is that Gender 1 individuals of high ability are willing to take on the risk of discrimination and apply for the discriminatory department. This creates a higher proportion of high ability G1s compared to G2s in that department. High ability compensates for discrimination and hides the discriminatory effect.
\(~\)
Sensitivity analysis
\(~\)
What are the implications of what we don’t know?
Assume confound exists, model its consequences for different strengths/kinds of influences
How strong must the confound be to change conclusions
Wow
You can vary the effect size of the confound to see what effect this has on the posterior
This allows you to get a posterior estimate of an effect while controlling for an assumed confound. You just need to estimate a reasonable effect size for the confound.
\(~\)
Fit a model with u added. Constrain the effect of u to be positive with a prior i.e. high ability only can have a positive effect on acceptance.
Population size and contact with other populations increase innovation.
The number of tools increases the likelihood of losing tools
DAG time!
Code
dag_coords <-tibble(name =c("C", "P", "L", "T"),x =c(1, 1, 2, 2),y =c(2, 1, 2, 1))dagify(C ~ L + P, P ~ L, T ~ C + P + L,coords = dag_coords) %>%gg_simple_dag()
Our goal is to estimate the effect of population size on tools.
Let’s identify all of the paths:
P -> T - front door path
P -> C -> T - C is a competing cause of tool count
P -> C <- L -> T
P <- L -> T - L creates a fork. This is a back-door path
\(~\)
Modelling tools - the poisson
\(~\)
Tool count is not binomial: this is because there is no maximum
We can instead use the poisson distribution
Very high maximum and very low probability of each success
For example, in these data there are many possible technologies, but very few are realised in any one place
The link function for the poisson distribution is the log link
This link function enforces positivity, as needed for a count
Note that this creates exponential scaling
Priors
If we continue our convention of using a Normal prior on the parameters, we should recognize those will be log-Normal distributed on the outcome scale. Why? Because we’re modelling the rate with the log link.
Lets plot an alpha prior of normal(0, 10) and one of Normal(3, 0.5)
Code
tibble(x =c(3, 22),y =c(0.055, 0.04),meanlog =c(0, 3),sdlog =c(10, 0.5)) %>%expand(nesting(x, y, meanlog, sdlog),number =seq(from =0, to =100, length.out =200)) %>%mutate(density =dlnorm(number, meanlog, sdlog),group =str_c("alpha%~%Normal(", meanlog, ", ", sdlog, ")")) %>%ggplot(aes(fill = group, color = group)) +geom_area(aes(x = number, y = density),alpha =3/4, size =0, position ="identity") +geom_text(data = . %>%group_by(group) %>%slice(1),aes(x = x, y = y, label = group),family ="Times", parse = T, hjust =0) +scale_fill_manual(values =met.brewer("Hokusai3")[1:2]) +scale_color_manual(values =met.brewer("Hokusai3")[1:2]) +scale_y_continuous(NULL, breaks =NULL) +xlab("mean number of tools") +theme_tidybayes() +theme(legend.position ="none")
Note the very large right tail for Normal (0, 10)
The second prior always expects some tools, which is more sensible for human societies
**How about *
Plot ~ Normal (0, 1) and ~ Normal (0, 10).
Also plot ~ Normal (3, 0.5) and ~ Normal (0, 0.2)
Code
set.seed(11)# how many lines would you like?n <-100# simulate and wrangletibble(i =1:n,a =rnorm(n, mean =3, sd =0.5)) %>%mutate(`beta%~%Normal(0*', '*10)`=rnorm(n, mean =0 , sd =10),`beta%~%Normal(0*', '*0.2)`=rnorm(n, mean =0 , sd =0.2)) %>%pivot_longer(contains("beta"),values_to ="b",names_to ="prior") %>%expand(nesting(i, a, b, prior),x =seq(from =-2, to =2, length.out =100)) %>%# plotggplot(aes(x = x, y =exp(a + b * x), group = i)) +geom_line(size =1/4, alpha =2/3,color =met.brewer("Hokusai3")[4]) +labs(x ="log population (std)",y ="total tools") +coord_cartesian(ylim =c(0, 100)) +facet_wrap(~ prior, labeller = label_parsed) +theme_tidybayes()
The right prior is truly awful - counts shoot into space! The left one is reasonable though.
Now we’ve settled on some sensible priors, let’s fit models
Code
# fit intercept modelOceanic_tools_1 <-brm(total_tools ~1,data = Oceanic_tools, family = poisson,prior =c(prior(normal(3, 0.5), class = Intercept)),iter =4000, warmup =2000, cores =4, chains =4,file ="fits/Oceanic_tools_1")# Fit interaction modelOceanic_tools_2 <-brm(total_tools ~1+ contact_id * log_pop_std,data = Oceanic_tools, family = poisson,prior =c(prior(normal(3, 0.5), class = Intercept),prior(normal(0, 0.2), class = b)),iter =4000, warmup =2000, cores =4, chains =4,file ="fits/Oceanic_tools_2")
Hawaii in particular is having a huge effect. Note also that the high contact islands are not represented past a certain population size, but the model continues to predict. This has huge uncertainty. Another quirk of the model is that it predicts a non-zero number of tools for islands with 0 people. This is nonsense.
Two immediate ways to improve model
Use a model robust to outliers: for this distribution type we can use the gamma-poisson (negative binomial)
Use a more principled scientific model
We can model this differently:
We can model the change per unit time.
\(\Delta T = \alpha P^\beta - \gamma T\)
Where
T is the change in tools
is the innovation rate
establishes diminishing returns
then subtract T, the rate of loss
This is not a regression model.
If we want to find the expected number of tools for a certain population size, we simply solve for T.
\(T = \frac{\alpha_{c}P^{\beta C}}{\gamma}\)
Sub this in for in our poisson model!
Lets fit the innovation/loss model
All parameters are rates, so they must be positive.
Two options here:
use exp()
use appropriate prior to constrain it to be positive